Introducción
Durante décadas, el abordaje clínico de la enfermedad de Alzheimer (EA) ha estado marcado por un palpable nihilismo terapéutico, limitado al tratamiento de los síntomas mediante inhibidores de la colinesterasa y memantina, sin capacidad real para alterar el curso biológico de la neurodegeneración. Sin embargo, el panorama actual refleja un cambio de paradigma hacia la era de las terapias modificadoras de la enfermedad (DMT, por sus siglas en inglés). Este giro se fundamenta en la validación clínica, al menos en parte, de la hipótesis de la cascada amiloide, materializada en la llegada de anticuerpos monoclonales dirigidos contra las formas agregadas del péptido beta-amiloide1.
Este hito histórico no ha estado exento de controversia. Se inició con la aprobación acelerada y discutida de aducanumab en EE.UU., pero se ha consolidado recientemente con la evidencia robusta de ensayos de fase III para lecanemab2 y donanemab3. Ambos fármacos han demostrado no solo una reducción significativa de la carga de amiloide cerebral, sino, de manera crucial, una ralentización del deterioro cognitivo y funcional en estadios tempranos de la enfermedad4.
No obstante, la tesis central que nos ocupa trasciende la eficacia farmacológica per se. El verdadero reto que afrontamos reside en la idoneidad y resiliencia de nuestro sistema sanitario para absorber esta innovación. Como se ha evidenciado en encuestas a responsables de los servicios de neurología en España, la implementación de estas terapias exigirá cambios estructurales profundos en nuestros departamentos, desde la gestión de recursos humanos hasta la disponibilidad de técnicas de neuroimagen y biomarcadores específicos5.
Nos encontramos, por tanto, ante lo que podemos definir como una «triple revolución». En primer lugar, una revolución diagnóstica, que exige la transición definitiva hacia un diagnóstico biológico de precisión mediante biomarcadores en líquido cefalorraquídeo (LCR) o PET-amiloide, y potencialmente en plasma en el futuro cercano6. En segundo lugar, una revolución terapéutica inmediata, que implica la administración de tratamientos intravenosos complejos con un perfil de seguridad que requiere monitorización estricta de las anomalías de imagen relacionadas con el amiloide (ARIA)7. Y finalmente, una revolución asistencial, quizás la más desconocida y desafiante, que nos obliga a redefinir los flujos de pacientes y la coordinación entre atención primaria y especializada para evitar el colapso de las unidades de memoria8.
Mecanismos de acción y evidencia clínica
La hipótesis del amiloide revisada
La reciente validación clínica de los anticuerpos monoclonales antiamiloide no debe interpretarse simplemente como una confirmación tardía de la hipótesis de la cascada amiloide, formulada hace tres décadas, sino como el refinamiento de su aplicación farmacológica. El fracaso de generaciones previas de fármacos no refutó la patogenicidad del amiloide, sino que evidenció la necesidad de una mayor especificidad y potencia de depuración. La clave del éxito actual reside en la alta afinidad de estas nuevas moléculas por conformaciones específicas del péptido y su enorme potencial para eliminarlo.
El lecanemab, por ejemplo, es un anticuerpo humanizado IgG1 que se une con alta selectividad a las protofibrillas solubles de beta-amiloide, especies que se consideran neurotóxicas y que impulsan la fisiopatología de la enfermedad antes y después de la formación de placas9. Por su parte, el donanemab se dirige contra una forma truncada y piroglutamada del péptido (N3pG) presente exclusivamente en las placas amiloides insolubles, facilitando una eliminación masiva mediante fagocitosis mediada por la microglía3,9 (Fig. 1).
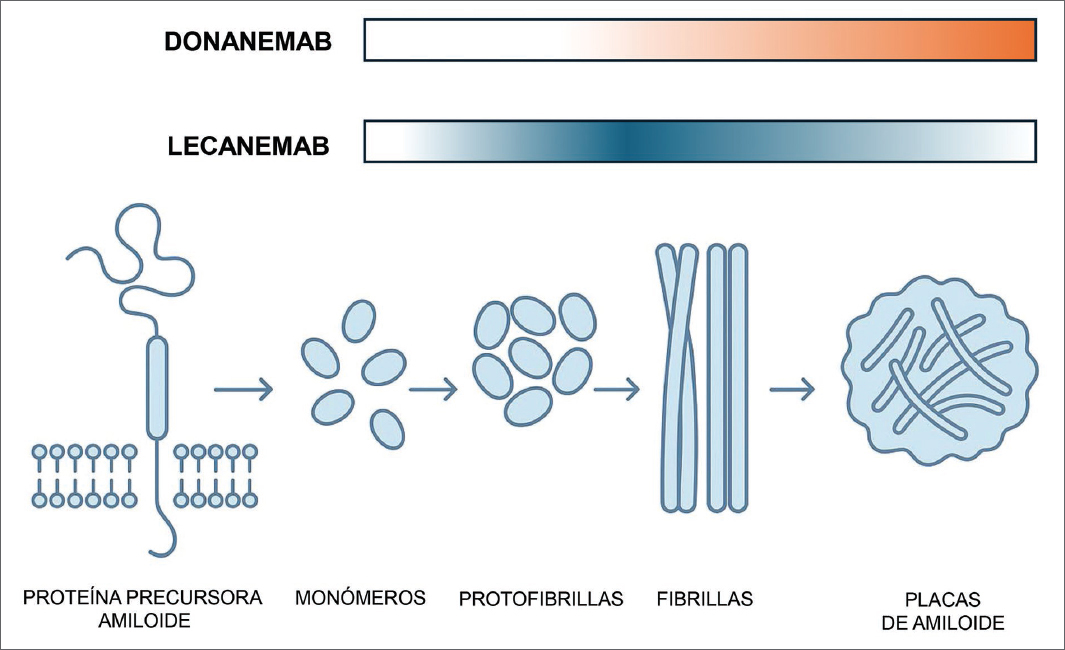
Figura 1. Esquema de las dianas terapéuticas de ambos anticuerpos antiamiloide. El lecanemab tiene una afinidad relevante frente a las protofibrillas y fibrillas de beta-amiloide, mientras que el donanemab presenta alta afinidad sobre el beta-amiloide de las placas.
A diferencia de sus predecesores, estos agentes logran reducciones profundas de la carga amiloide, llevando a la mayoría de los pacientes a niveles de «negatividad de amiloide» en las pruebas de tomografía por emisión de positrones (PET). Sin embargo, desde una perspectiva neurobiológica, debemos ser cautos; aunque la eliminación del amiloide ralentiza el declive, no detiene la enfermedad. Los procesos patológicos vinculados con el Alzheimer son multifacéticos e involucran tauopatía, neuroinflamación y disfunción sináptica, que pueden perpetuarse independientemente de la depuración del amiloide, especialmente en estadios avanzados1,10. Por tanto, estos fármacos representan un hito necesario, pero insuficiente como monoterapia en la búsqueda de una curación completa.
Análisis de los ensayos pivotales
La evidencia que sustenta este cambio de paradigma proviene de dos ensayos de fase III con diseños rigurosos. El estudio Clarity AD evaluó el lecanemab en 1.795 participantes con deterioro cognitivo leve (DCL) o demencia leve. Tras 18 meses, el fármaco demostró una reducción del 27% en el declive clínico medido por la escala Clinical Dementia Rating-Sum of Boxes (CDR-SB), el objetivo primario, acompañado de una reducción significativa de la carga amiloide2.
Paralelamente, el estudio TRAILBLAZER-ALZ 2 con donanemab y un número de participantes similar (n = 1.736) introdujo un diseño innovador estratificado por niveles de proteína tau. En la población con niveles bajos/medios de tau, el donanemab ralentizó el declive en un 35% en la escala iADRS (Integrated Alzheimer’s Disease Rating Scale) y en un 36% en la CDR-SB a las 76 semanas3. Un aspecto distintivo de este ensayo fue su enfoque de «tratar hasta aclarar». El tratamiento se detuvo cuando las concentraciones de amiloide en PET descendieron por debajo de un umbral predefinido, lo cual ocurrió en aproximadamente el 80% de los pacientes al año y medio.
Magnitud del efecto: la traducción a la vida real
Como clínicos, la pregunta fundamental no es la significación estadística, sino la relevancia clínica: ¿qué implica para un paciente y su familia «frenar un 27-35%» el deterioro? En términos tangibles, esta ralentización se traduce en un ahorro de tiempo. Los análisis de los ensayos sugieren que el tratamiento proporciona un retraso en la progresión de la enfermedad de aproximadamente 5 a 7 meses durante un periodo de tratamiento de 18 meses3,11.
Esto significa prolongar la fase de la enfermedad en la que el paciente mantiene su autonomía, su capacidad para reconocer a sus familiares y realizar actividades instrumentales de la vida diaria. Aunque el efecto absoluto en las escalas psicométricas (p. ej., 0,45 puntos en CDR-SB para lecanemab) ha sido objeto de debate por situarse en los márgenes de la diferencia mínima clínicamente importante, el beneficio acumulativo de preservar la función en estadios tempranos parece claro desde la perspectiva del paciente y del cuidador. Además, se observó un menor riesgo de progresión al siguiente estadio de gravedad de la demencia1,2,10.
La ventana de oportunidad y el horizonte a largo plazo
La eficacia de estas terapias está intrínsecamente ligada a la «ventana de oportunidad» terapéutica al estar indicado en la EA prodrómica y la demencia leve asociada a EA. Los datos sugieren que la eficacia disminuye a medida que avanza la patología tau y el deterioro cognitivo; de hecho, en análisis de subgrupos, los pacientes con niveles altos de tau o estadios más avanzados mostraron menor beneficio10. Esto refuerza la necesidad imperiosa de diagnosticar biológicamente la enfermedad en sus fases más incipientes si queremos ser eficaces y eficientes en la implementación de estas terapias12.
A largo plazo, persisten incertidumbres que solo los estudios de extensión abierta y los registros de vida real podrán esclarecer. Hay incertidumbre en relación con si el beneficio clínico diverge y aumenta con el tiempo más allá de los 18 meses o si se estabiliza13. Todavía no disponemos de datos publicados sobre la extensión abierta del ensayo y sus resultados en práctica clínica real. No obstante, datos reportados por la propia compañía que comparan la evolución del grupo tratado con una cohorte natural obtenida de la base de datos ADNI establece diferencias crecientes a lo largo del tiempo. En un análisis a 10 años, los pacientes tratados tardaron 2,5 años más en progresar desde DCL a demencia leve por Alzheimer (9,7 vs. 7,2 años en el grupo sin tratamiento)14. En cuanto al donanemab, los datos presentados por la compañía (sin publicar) presenta resultados similares. Tras 3 años, los participantes tratados con donanemab mostraron una ralentización del declive de entre el 39 y el 48% en las escalas iADRS y CDR-SB en comparación con la cohorte de ADNI. La diferencia absoluta en la escala CDR-SB fue de 1,21 puntos a favor de donanemab a los 36 meses, claramente superior a los 0,67 puntos reportados a los 18 meses en el ensayo clínico15.
Asimismo, el modelo del donanemab plantea la posibilidad de tratamientos finitos, similares a la inducción en oncología, donde se suspende la terapia tras limpiar el cerebro de amiloide, reiniciándose solo si reaparece la enfermedad3. Esta estrategia podría tener profundas implicaciones en la sostenibilidad financiera y la carga logística para los sistemas de salud10. En todo caso, hemos pasado de la futilidad a la posibilidad, pero la gestión de las expectativas y la selección precisa del paciente serán las herramientas más valiosas del neurólogo en esta nueva era (Tabla 1).
Tabla 1. Anticuerpos aprobados por la EMA para uso en enfermedad de Alzheimer
| Característica | Lecanemab | Donanemab |
|---|---|---|
| Mecanismo biológico | Anticuerpo IgG1 que reconoce específicamente formas agregadas solubles (protofibrillas) e insolubles de beta-amiloide, reduciendo las placas | Anticuerpo IgG1 con alta afinidad por una forma modificada de beta-amiloide truncada (N3pE Ab) presente en las placas; elimina las placas mediante fagocitosis mediada por microglía |
| Posología (dosis) | Dosis fija: 10 mg/kg de peso corporal | Dosis escalonada: 350 mg (1ª dosis), 700 mg (2ª dosis), 1.050 mg (3ª dosis) y 1.400 mg (mantenimiento) |
| Frecuencia | Una vez cada 2 semanas | Una vez cada 4 semanas |
| Forma de administración | Perfusión iv durante aproximadamente 1 hora | Perfusión iv durante al menos 30 minutos |
| Duración del tratamiento | Debe interrumpirse permanentemente si el paciente progresa a enfermedad de demencia moderada | Se mantiene hasta confirmar el aclaramiento de placas por método validado; duración máxima de 18 meses (incluso si no hay aclaramiento) |
| Monitorización (RM) | RM basal (6 meses previos) | RM basal (6 meses previos) |
| RM de seguimiento antes de las perfusiones 3ª, 5ª, 7ª y 14ª | RM antes de la 2ª, 3ª, 4ª y 7ª dosis (meses 1, 2, 3 y 6) | |
| Una extra al año en determinadas circunstancias de riesgo | ||
| Perfil genético (ApoE e4) | Indicado para no portadores o heterocigotos Contraindicado en homocigotos | Indicado para no portadores o heterocigotos Contraindicado en homocigotos |
| Observación posperfusión | 2,5 horas tras la primera perfusión para detectar reacciones | Mínimo 30 minutos después de cada perfusión |
|
Diferencias en el uso de los dos anticuerpos antiamiloide según las características específicas aprobadas por la EMA en ficha técnica. EMA: European Medicines Agency; IgG1: inmunoglobulina G1; iv: intravenosa; RM: resonancia magnética. |
||
El desafío del diagnóstico de precisión en el Sistema Nacional de Salud
La llegada de los anticuerpos monoclonales antiamiloide no solo supone una novedad terapéutica, sino que obliga a una reingeniería completa de nuestros procesos asistenciales. Hasta ahora, el diagnóstico de la EA en la práctica clínica habitual ha sido fundamentalmente fenotípico, apoyado en la exclusión de otras causas. Sin embargo, la prescripción de fármacos como lecanemab o donanemab exige una certeza biológica. El paso de un diagnóstico de probabilidad a un diagnóstico de precisión molecular representa el mayor desafío logístico al que se enfrenta el Sistema Nacional de Salud (SNS) en el ámbito de la neurología cognitiva.
El papel de la atención primaria en el diagnóstico
Un análisis europeo de la capacidad de atención primaria en el diagnóstico de EA muestra que una mayoría sustancial de los médicos de atención primaria (87,8%) se autopercibe con la capacidad y el conocimiento necesarios para diagnosticar la enfermedad en estadios tempranos. No obstante, estos datos muestran una discrepancia significativa respecto a la visión de los especialistas, quienes identifican una insuficiencia formativa y una carencia crítica de herramientas diagnósticas avanzadas en este primer nivel asistencial. Esta discrepancia en recursos y formación genera una inercia terapéutica evidente: el médico de atención primaria, al carecer de acceso directo a biomarcadores y de guías estandarizadas, inicia tratamiento farmacológico en solo un 33,7% de los casos de demencia leve, frente al 48,5% en atención especializada. Esta barrera sistémica, sumada a una demora media de 15,2 meses desde el inicio de los síntomas hasta la primera consulta, provoca que el paciente llegue al diagnóstico final casi tres años tarde, cerrando la ventana de oportunidad clínica necesaria para la eficacia de los fármacos modificadores de la enfermedad16.
Criterios de elegibilidad: la selección del paciente adecuado
Los criterios de elegibilidad definidos en los ensayos clínicos y reflejados en las fichas técnicas y recomendaciones europeas son estrictos y excluyentes. El tratamiento está indicado exclusivamente para pacientes con DCL o demencia leve debida a la enfermedad de Alzheimer, con confirmación de patología amiloide2,3.
En el contexto europeo y español, la selección del paciente se vuelve aún más restrictiva por motivos de seguridad. Las recomendaciones de uso apropiado y las decisiones regulatorias recientes subrayan la necesidad de excluir a pacientes con mayor riesgo de ARIA. Específicamente, la Agencia Europea de Medicamentos (EMA) ha puesto el foco en la exclusión de pacientes anticoagulados y, de manera crucial, se ha establecido la no elegibilidad de los portadores homocigotos de ApoE e4 en las fichas técnicas aprobadas, dado su elevado riesgo de ARIA-E o ARIA-H17–19.
Además, la elegibilidad requiere una evaluación de resonancia magnética (RM) basal rigurosa para descartar patología cerebrovascular significativa (definida comúnmente con un grado Fazekas III), presencia de más de cuatro microhemorragias, siderosis superficial o una hemorragia previa mayor de 1 cm. Por tanto, la ecuación clínica para iniciar tratamiento no es solo síntomas más amiloide, sino una compleja valoración de estadio clínico precoz, carga amiloide positiva, perfil genético de riesgo bajo/moderado y ausencia de angiopatía amiloide cerebral relevante18,19.
Biomarcadores: la dicotomía PET-amiloide vs. líquido cefalorraquídeo
Para cumplir con el requisito de confirmar la patología amiloide, el SNS debe resolver la tensión entre la disponibilidad y la demanda. Actualmente, disponemos de dos vías validadas: la PET de amiloide y el análisis de biomarcadores (amiloide beta42, amiloide beta40, p-tau, t-tau) en LCR7.
En España, la disponibilidad de equipos PET y, más críticamente, de trazadores amiloides, es heterogénea y su coste es elevado. Aunque la PET es menos invasiva y preferida por los pacientes, la realidad logística de nuestros hospitales de segundo y tercer nivel sugiere que el análisis de LCR mediante punción lumbar (PL) seguirá siendo la «piedra angular» del diagnóstico biológico a corto plazo. Los responsables de los servicios de neurología españoles coinciden en que la implementación de estas terapias requerirá un aumento significativo en la capacidad de realizar punciones lumbares y en la contratación de especialistas en análisis para el procesamiento de muestras, dado que la capacidad actual de los servicios de medicina nuclear es insuficiente para absorber la demanda proyectada de PET-amiloide como herramienta de cribado masivo5.
El papel emergente de los biomarcadores en sangre: p-tau 217
Ante el «cuello de botella» que suponen la PET y la PL, los biomarcadores plasmáticos emergen como la solución más prometedora para la sostenibilidad del sistema. Específicamente, la fosfo-tau 217 (ptau217) en plasma ha demostrado una precisión diagnóstica equiparable a los biomarcadores en LCR para identificar patología amiloide cerebral20. Su papel inmediato en la atención especializada debe ser el de triaje o cribado de alta precisión. La implementación de un protocolo de biomarcadores en sangre permitiría descartar con una alta fiabilidad a aquellos pacientes con deterioro cognitivo que no presentan amiloidosis cerebral, evitando así la realización de pruebas invasivas o costosas (PET/LCR). Se estima que el uso de ptau217 podría reducir la necesidad de pruebas confirmatorias complejas en un porcentaje muy significativo de la población consultante12,21.
Mirando hacia el futuro, la validación de estos marcadores en atención primaria podría transformar radicalmente el flujo de pacientes. Estudios recientes indican que la precisión diagnóstica de los médicos de atención primaria utilizando ptau217 es significativamente superior a la evaluación clínica estándar. Sin embargo, hasta que estas determinaciones estén plenamente estandarizadas y comercializadas, su uso debe circunscribirse a unidades de memoria para preseleccionar candidatos a tratamiento confirmatorio5,20,22.
Infraestructura de neurología: saturación vs. especialización
La infraestructura actual de los servicios de neurología en España no está dimensionada para asumir la carga asistencial que implican los nuevos fármacos. La «consulta de neurología general» con tiempos de visita limitados es incompatible con la complejidad del proceso que incluye selección clínica, discusión de riesgos/beneficios, gestión de biomarcadores, genotipado ApoE, programación de infusiones quincenales o mensuales y monitorización de seguridad mediante RM seriada para detectar ARIA5,11. Por todo ello, es imperativa la creación y dotación de unidades de memoria especializadas o el refuerzo de las existentes. Estas unidades pueden operar bajo modelos de hub and spoke (red de centros), donde centros expertos (hubs) gestionen la indicación, infusión y monitorización compleja, apoyados por centros locales (spokes) para el cribado inicial y seguimiento8.
La encuesta a líderes de neurología en España revela un consenso absoluto (100% de acuerdo) sobre la necesidad de sistematizar la atención en unidades específicas y aumentar la contratación no solo de neurólogos, sino de neuropsicólogos y enfermería de práctica avanzada (con rol de gestoras de casos)5. Sin una inversión decidida en recursos humanos y estructurales (hospitales de día, tiempo de RM reservado, personal de enfermería), el riesgo no es solo la incapacidad de administrar el fármaco, sino el colapso de la atención al resto de afecciones neurológicas por el desplazamiento de recursos. El diagnóstico de precisión es, por tanto, un desafío organizativo y científico.
Seguridad y manejo de las complicaciones potenciales
La implementación clínica de estas nuevas terapias conlleva un cambio sustancial en la farmacovigilancia. A diferencia de los tratamientos sintomáticos convencionales, estas terapias biológicas presentan un perfil de seguridad complejo, dominado por las ARIA.
ARIA-E y ARIA-H: fenomenología y estratificación del riesgo
Las ARIA representan la manifestación radiológica de la alteración de la permeabilidad vascular inducida por la eliminación de los depósitos de amiloide en los vasos cerebrales. Se clasifican en dos entidades, las ARIA-E, caracterizadas por edema vasogénico o efusiones sulcales, y las ARIA-H, definidas por microhemorragias y siderosis superficial19.
La incidencia no es despreciable. En el ensayo Clarity AD de lecanemab se observaron ARIA en el 21,5% de los pacientes tratados, siendo el 12,6% ARIA-E y el 17,3% ARIA-H2. Con donanemab las tasas fueron superiores, alcanzando un 36,8% de ARIA global, con un 24% de ARIA-E y un 31,4% de ARIA-H3. Aunque la mayoría de estos eventos son asintomáticos (aproximadamente el 80%), cuando presentan síntomas, estos pueden oscilar desde cefaleas leves y confusión hasta convulsiones o déficits focales graves1,17.
El factor de riesgo más determinante es el genotipo ApoE. Existe un efecto de dosis génica claro, de forma que los portadores homocigotos de ApoE e4 presentan un riesgo drásticamente mayor de sufrir ARIA (hasta un 45-55% de incidencia), así como formas más graves y recurrentes en comparación con los no portadores o heterocigotos. Esta evidencia ha llevado a la EMA a restringir la indicación de lecanemab y donanemab exclusivamente a pacientes no portadores o heterocigotos de ApoE e4, excluyendo a los homocigotos por un balance beneficio-riesgo desfavorable18,19.
El desafío del protocolo de imagen
La seguridad del paciente depende de la detección precoz de las potenciales complicaciones. Los protocolos de las fichas técnicas y las recomendaciones de uso apropiado exigen una RM basal reciente (menos de 6-12 meses) para descartar enfermedad vascular. Una vez iniciado el tratamiento, la carga de monitorización es intensa. Para lecanemab, se requieren RM de seguridad antes de las infusiones 3ª, 5ª, 7ª y 14ª (aproximadamente a los 1, 2, 3 y 6 meses), coincidiendo con la ventana temporal de mayor riesgo de aparición de ARIA. El donanemab exige un esquema similar, con RM previas a la 2ª, 3ª, 4ª y 7ª dosis (aproximadamente a los 2, 3, 4 y 7 meses). En este fármaco se debería realizar otra RM al año de tratamiento si existe antecedente de ARIA o el paciente es heterocigoto ApoE e412,17–19.
Aquí surge una pregunta crítica de gestión sanitaria: ¿dispone el SNS de suficiente disponibilidad de RM para asumir este seguimiento estricto? La realidad actual de las listas de espera sugiere que la demanda de RM podría convertirse en uno de los cuellos de botella para la implementación4. Para evitarlo, los expertos recomiendan optimizar los recursos mediante protocolos de RM abreviados (aproximadamente 10-15 minutos) que incluyan exclusivamente secuencias FLAIR (para ARIA-E) y T2-eco gradiente o susceptibilidad magnética (SWI) para ARIA-H, evitando otras secuencias innecesarias. Además, sería imprescindible garantizar el acceso a RM urgente para diferenciar un evento ARIA sintomático de un ictus isquémico, dado que el uso de trombolíticos en un paciente con ARIA podría ser fatal9,10,17,23.
Manejo clínico: suspensión, reintroducción y el paciente informado
El manejo de las ARIA se basa en algoritmos de decisión que cruzan la gravedad radiológica con la sintomatología clínica del paciente que sufre dicha complicación (Tablas 2 y 3).
Tabla 2. Manejo comparativo de ARIA-E (edema/efusión)
| Gravedad radiológica | Gravedad clínica | Manejo lecanemab | Manejo donanemab |
|---|---|---|---|
| Leve (< 5 cm) | Asintomático | Puede continuar la administración | Valorar suspender temporalmente según imagen y clínica |
| Moderada (5-10 cm) | Asintomático | Suspender temporalmente | Suspender temporalmente |
| Grave (> 10 cm) | Asintomático | Suspender temporalmente | Interrumpir permanentemente |
| Leve/moderada | Sintomático | Suspender temporalmente | Suspender temporalmente |
| Grave | Sintomático | Suspender temporalmente, pero interrumpir si es la 2ª vez | Interrumpir permanentemente |
|
Tabla resumen del manejo de las ARIA-E durante el tratamiento con lecanemab o donanemab según ficha técnica. Las ARIA-E se monitorizan mediante la secuencia RM-FLAIR para observar el edema cerebral o efusiones en los surcos. ARIA-E: Amyloid-Related Imaging Abnormalities-Edema o Anomalías de Imagen Relacionadas con el Amiloide-Edema o Efusión; RM: resonancia magnética; FLAIR: Fluid-Attenuated Inversion Recovery. |
|||
Tabla 3. Manejo comparativo de ARIA-H (microhemorragias y siderosis)
| Gravedad radiológica | Gravedad clínica | Manejo lecanemab | Manejo donanemab |
|---|---|---|---|
| Leve (≤ 4 microH) | Asintomático | Puede continuar la administración | Valorar suspender temporalmente |
| Moderada (5 a 9 microH) | Asintomático | Suspender temporalmente | Suspender temporalmente |
| Grave (≥ 10 microH) | Asintomático | Interrumpir permanentemente | Interrumpir permanentemente |
| Leve/moderada | Sintomático | Suspender temporalmente | Suspender temporalmente |
| Grave | Sintomático | Interrumpir permanentemente | Interrumpir permanentemente |
| Hemorragia > 1 cm | Cualquier situación | Interrumpir permanentemente | Interrumpir permanentemente |
|
Tabla resumen del manejo de las ARIA-H durante el tratamiento con lecanemab o donanemab según ficha técnica. El manejo de las ARIA-H depende del número de nuevas microhemorragias y áreas de siderosis superficial detectadas en la nueva RM. Se monitorizan mediante la secuencia eco-gradiente T2 o imagen ponderada por susceptibilidad (SWI). ARIA-H: Amyloid-Related Imaging Abnormalities-Hemorrhage o Anomalías de Imagen Relacionadas con el Amiloide-Hemorragia o depósito de hemosiderina; microH: microhemorragias; RM: resonancia magnética. |
|||
Aunque el manejo de ambos fármacos es similar, existen algunas diferencias que manifestar. Una de las diferencias críticas radica en el umbral de interrupción definitiva del tratamiento; el donanemab adopta una postura más estricta al prescribir la interrupción permanente inmediata ante la primera aparición de un evento de ARIA-E radiográficamente grave. En contraste, el lecanemab permite un manejo más flexible, autorizando la suspensión temporal y la posterior reanudación una vez se confirme la resolución del primer evento grave, reservando la interrupción definitiva para una segunda recurrencia de un cuadro grave. Esta disparidad en la tolerancia clínica también se observa en el manejo de los casos leves asintomáticos. Mientras que el protocolo del lecanemab indica que la administración puede continuar de forma directa en estas situaciones, el donanemab insta al médico a valorar la suspensión temporal, condicionando la continuidad a una evaluación rigurosa de la estabilidad clínica y radiológica. Finalmente, en lo que respecta al tratamiento de soporte, la ficha técnica del donanemab es la única que menciona explícitamente el uso de corticosteroides como una opción terapéutica para el manejo de ARIA-E, aunque subraya que su eficacia clínica aún no ha sido formalmente establecida.
Finalmente, la gestión de expectativas y la seguridad clínica pasan por un proceso de consentimiento informado robusto. No se trata de un trámite burocrático, sino de una educación sanitaria activa. La EMA advierte que el paciente debe ser portador de una «tarjeta de alerta del paciente» que indique a cualquier médico de urgencias sobre su tratamiento, así como la contraindicación de la administración de trombolíticos o anticoagulantes sin una valoración detallada previa. Asimismo, el consejo genético previo a la prueba de ApoE es mandatorio, debiendo explicar las implicaciones no solo para la elegibilidad del tratamiento, sino para el riesgo de sus descendientes. En definitiva, la seguridad de estas terapias no reside solo en el fármaco, sino en la solidez de la estructura asistencial que lo rodea y que es clave para que el programa tenga éxito11,13,17.
Implementación y sostenibilidad en España
Tras la evaluación de la EMA que ha recomendado la autorización de comercialización de lecanemab y donanemab para una población restringida, el foco se desplaza ahora hacia la sostenibilidad financiera. Es previsible que la autorización nacional venga acompañada de protocolos farmacoclínicos que limiten la prescripción a centros de referencia siguiendo modelos de acceso controlado para garantizar la seguridad del paciente y la viabilidad del sistema7,13.
El debate económico no debe centrarse exclusivamente en el precio de adquisición del fármaco. La implementación de estas terapias conlleva una carga de costes indirectos o estructurales que podría superar al coste farmacológico directo. Como señalan Matias-Guiu et al.5, la absorción de estas terapias exige una inversión en recursos humanos y técnicos como la contratación de enfermería, aumento de los puestos para la administración intravenosa en hospitales de día, ampliación de las plantillas de neurólogos y neuropsicólogos, y, de forma crítica, el coste derivado de la realización de las RM de seguridad. A esto se suma el coste de los biomarcadores (PET o LCR) y el genotipado ApoE, imprescindibles para la elegibilidad. Sin una inyección presupuestaria específica que contemple estos costes ocultos, la prescripción del fármaco será inviable en la práctica real, independientemente de su aprobación administrativa4,9.
La heterogeneidad del SNS, con 17 servicios de salud autonómicos, plantea un riesgo tangible de inequidad. Existe el peligro de fracturar la atención a la demencia en una «España a dos velocidades»: una con acceso a grandes hospitales terciarios dotados de unidades de memoria, PET y guardias 24/7 capaces de gestionar complicaciones como ARIA; y otra, en áreas rurales u hospitales comarcales, donde la infraestructura es insuficiente para cumplir los requisitos de la ficha técnica. Para mitigar estas disparidades, es imperativo establecer redes asistenciales de tipo hub and spoke que garanticen que el código postal del paciente no determine su elegibilidad para un DMT5,7,8.
Finalmente, nos enfrentamos a un desafío ético en la consulta. La llegada de estos fármacos ha generado unas expectativas sociales que, a menudo, no se alinean con los estrictos criterios de inclusión. Tendremos que gestionar la frustración de familias con pacientes en estadios moderados o avanzados que quedan fuera de la indicación terapéutica, así como comunicar a pacientes asintomáticos o con deterioro leve que su perfil genético (homocigosis ApoE4) o sus comorbilidades (anticoagulación) les excluyen del tratamiento por motivos de seguridad. La selección del paciente debe regirse por la beneficencia y la no maleficencia, evitando la «medicina de la esperanza» infundada. El consentimiento informado deberá ser un proceso educativo, donde se discuta transparentemente que el objetivo es la ralentización del declive, no la curación, y se asuman riesgos graves potenciales por un beneficio clínico modesto11–13.
¿Estamos preparados? La ruta de implementación de la neurología española
La incorporación de los anticuerpos monoclonales antiamiloide al arsenal terapéutico exige una transformación profunda de la estructura asistencial de la neurología en España. No estamos ante una simple adición farmacológica, sino ante un cambio de modelo que requiere pasar de una atención fragmentada a procesos de alta integración. Para que esta transición sea exitosa y segura, debemos actuar sobre tres ejes fundamentales: la capacitación profesional, la reingeniería de los circuitos asistenciales y la generación de evidencia en vida real.
Formación: capacitación más allá de la unidad de demencia
Si bien el manejo directo de estas terapias recaerá en las unidades de memoria, la magnitud de la demanda asistencial prevista hace insostenible que el conocimiento quede confinado exclusivamente en los subespecialistas en neurología cognitiva y de la conducta. Es imperativo desplegar programas de capacitación para neurólogos generales y residentes, enfocados en la identificación precisa de candidatos y, críticamente, en la detección de complicaciones. Como señalan Matias-Guiu et al.5, existe un consenso del 93,75% entre los responsables de los servicios españoles sobre la necesidad de una formación específica que habilite a los neurólogos para integrarse en estas unidades o, al menos, para realizar un cribado eficaz que evite cuellos de botella en los centros de referencia. Asimismo, la formación debe extenderse a la neurorradiología para la estandarización en la lectura de ARIA y a los servicios de urgencias para el manejo de potenciales eventos agudos, evitando la iatrogenia17.
Circuitos asistenciales: el equipo multidisciplinar
La administración de estos nuevos fármacos requiere de una coordinación logística sin precedentes, similar a la observada en los códigos ictus o en la oncología médica. El neurólogo debe actuar como director de orquesta en un circuito que integre fluidamente a los siguientes colegas:
- – Radiología. Para garantizar la disponibilidad de RM basales y de seguridad en tiempos estrictos9.
- – Medicina nuclear y laboratorio. Para la confirmación de biomarcadores (PET o LCR) y el genotipado ApoE de forma ágil7.
- – Farmacia hospitalaria. Para la preparación y gestión de los biológicos11.
- – Enfermería de práctica avanzada. Esta figura se perfila como una de los elementos clave del proceso. La incorporación de enfermería gestora de casos es avalada por el 100% de los líderes de neurología en España para coordinar las infusiones, monitorizar efectos adversos y servir de enlace con el paciente5.
Registros de vida real: la vigilancia poscomercialización
Finalmente, la autorización regulatoria no cierra el capítulo de la evidencia científica. La EMA exige la implementación de registros de seguridad postautorización para caracterizar la incidencia de ARIA y la efectividad a largo plazo fuera del entorno controlado de los ensayos clínicos. España debe comprometerse con la creación o adaptación de bases de datos nacionales (como el registro ALZ-NET en EE.UU.) que permitan monitorizar no solo la seguridad, sino también la efectividad clínica en una población más heterogénea y con mayor comorbilidad que la de los ensayos pivotales. Solo mediante la recolección sistemática de datos en vida real podremos validar el impacto farmacoeconómico y ajustar nuestros protocolos clínicos para garantizar la sostenibilidad del sistema11.
Conclusiones
La aprobación de lecanemab y donanemab marca un punto de inflexión histórico, pero no constituye la solución definitiva a la compleja biología de la EA. Como comunidad científica, debemos asumir que el futuro inmediato transita hacia una medicina personalizada, donde el perfil biológico individual (presencia de amiloide, tau, otras proteinopatías, neuroinflamación) dictará el uso de terapias combinadas capaces de abordar la multifactorialidad de la enfermedad más allá de la cascada amiloide1,21. Los anticuerpos actuales son, probablemente, la «piedra angular» sobre la que se edificarán regímenes terapéuticos más potentes y seguros, posiblemente dirigidos a estadios presintomáticos13,23.
Los datos de preparación de los sistemas sanitarios europeos y el análisis específico de los servicios de neurología españoles revelan una brecha significativa entre la innovación farmacológica y la capacidad asistencial instalada4,5. Si bien hemos superado el nihilismo terapéutico, nos enfrentamos a cuellos de botella críticos en el diagnóstico por biomarcadores, la disponibilidad de RM y la dotación de recursos humanos especializados8,24. El éxito de estas terapias en el mundo real no dependerá únicamente de su eficacia molecular, sino de nuestra agilidad para transformar los modelos de gestión clínica, integrar eficazmente la atención primaria con la especializada y garantizar que el acceso a la innovación no genere nuevas inequidades en nuestro sistema de salud16.
Financiación
El presente trabajo no ha recibido ninguna subvención oficial, beca o apoyo de un programa de investigación destinados a la redacción de su contenido.
Conflicto de intereses
El autor ha participado en actividades docentes y consultoría de Lilly, Roche y Eisai/Biogen.
Consideraciones éticas
Protección de personas y animales. El autor declara que para este trabajo no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad, consentimiento informado y aprobación ética. El estudio no involucra datos personales de pacientes ni requiere aprobación ética. No se aplican las guías SAGER.
Declaración sobre el uso de inteligencia artificial. El autor declara que no utilizó ningún tipo de inteligencia artificial generativa para la redacción de este manuscrito.
