INTERÉS DE ESTA REVISIÓN Y METODOLOGÍA EMPLEADA
La arteritis de células gigantes (ACG), denominada también arteritis de la temporal o enfermedad de Horton, es una vasculitis sistémica de etiología desconocida, que afecta a vasos de mediano y gran calibre con una predilección especial por las ramas extracraneales de la arteria carótida y, en menor medida, por la aorta y troncos supraaórticos1.
La incidencia media anual de ACG en España se estima en 7,42 casos por cada 100.000 personas/año (IC95%: 6,57-8,27). Afecta fundamentalmente a mujeres con edad > 50 años, siendo el pico máximo de incidencia a los 70-80 años2.
El interés de esta revisión reside en la actualización de las evidencias disponibles sobre el diagnóstico y el tratamiento de la ACG en la práctica clínica habitual.
Se ha seguido la metodología PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-analysis)3. Los criterios de inclusión fueron los siguientes: a) publicación entre el 2000 y 2023; b) estudios empíricos; c) escritos en español, inglés o francés, y d) publicados en revistas académicas de revisión por pares.
Se ha tomado como referencia el abordaje diagnóstico y terapéutico elaborados por el American College of Rheumatology (ACR) (https://rheumatology.org/vasculitis-guideline). Se han considerado también los protocolos publicados sobre ACG por la European Alliance of Associations for Rheumatology (EULAR) (https://www.eular.org/recommendations-management).
MANIFESTACIONES CLÍNICAS DE LA ARTERITIS DE CÉLULAS GIGANTES
Las manifestaciones clínicas son diversas. A nivel sistémico destacan: febrícula, astenia, anorexia y pérdida de peso. A nivel músculo-esquelético: polimialgia reumática (coexistente en aproximadamente el 50% de los casos), artralgias, mialgias, artritis periférica y tenosinovitis de tendones extensores4.
Dentro las manifestaciones craneales, cabe destacar la cefalea (60-98%) de características migrañosas localizada a nivel del área fronto-parietal o retroauricular, también occipital o facial (si palpamos la arteria temporal suele estar engrosada con hipersensibilidad o dolor al tacto), se puede acompañar de hiperalgesia en cuero cabelludo y claudicación mandibular. En casos graves se puede asociar a necrosis de cuero cabelludo, e incluso de la mucosa oral y lengua. La pérdida de visión unilateral o bilateral es una de las complicaciones más grave de la arteritis de la temporal (debida a una obstrucción de la arteria oftálmica). Otra manifestación grave, aunque menos frecuente, son los accidentes cerebrovasculares de tipo isquémico (causa más frecuente de muerte precoz en ACG), con predominancia del territorio posterior y basilar. De hecho, debe investigarse esta etiología si se han descartado otras causas más habituales de ictus en ancianos5.
También pueden verse afectados otros territorios vasculares, a destacar la aortitis (45-63%) principalmente de arteria torácica, aneurismas (causa más frecuente de muerte tardía), disección aórtica, afectación estenótica de grandes y medianos vasos, cardiopatía isquémica y pericarditis, tos seca u odinofagia y formas pseudotumorales. Las complicaciones a nivel del sistema nervioso periférico son menos frecuentes, en torno al 10-15%. Se puede manifestar como mononeuritis múltiple, y más raramente, como polineuropatía sensitivo-motora simétrica distal.
CRITERIOS DE CLASIFICACIÓN
Se dispone de dos esquemas para clasificar la ACG: el del ACR de 1990 y el del ACR/EULAR de 2022.
Criterios de clasificación ACR 19906
Deben cumplirse al menos tres de cinco criterios:
- Edad > 50 años.
- Cefalea de reciente comienzo o de características distintas a las habituales.
- Arteria temporal alterada en la exploración física con dolor a la palpación o pulsos disminuidos no debidos a arterioesclerosis.
- Velocidad de sedimentación globular (VSG) > 50 mm/h.
- Biopsia de arterial temporal compatible: vasculitis caracterizada por un infiltrado inflamatorio con predominio de células mononucleares o inflamación granulomatosa, frecuentemente con células gigantes multinucleadas.
Nuevos criterios de clasificación ACR/EULAR 20227
Para alcanzar el umbral de clasificación se deben tener ≥ 6 puntos.
Requisito fundamental:
- Edad ≥ 50 años en el momento del diagnóstico.
Criterios clínicos adicionales:
- Rigidez matutina en hombros o cuello +2
- Pérdida súbita de visión +3
- Claudicación mandibular o lingual +2
- Cefalea temporal de nueva aparición +2
- Hipersensibilidad en cuero cabelludo +2
- Exploración anormal de la arteria temporal +2.
Criterios de laboratorio, imagen y biopsia:
- VSG ≥ 50 mm/h o proteína C reactiva (PCR) ≥ 10 mg/l +3
- Biopsia de arteria temporal positiva o signo del halo +5
- Afectación axilar bilateral +2
- Tomografía por emisión de positrones con fluorodesoxiglucosa (FDG-PET) con actividad a lo largo de la aorta +2.
PROTOCOLO DE ACTUACIÓN DIAGNÓSTICO Y TERAPÉUTICO A SEGUIR EN PACIENTES CON ARTERITIS DE CÉLULAS GIGANTES
Valoración inicial
A todo paciente con ACG se le debe realizar:
Historia clínica completa
Se debe insistir en los síntomas típicos de la enfermedad.
Exploración física completa por aparatos
Es necesario incidir en la exploración del sistema vascular, y especialmente, de las arterias temporales. Se debe explorar minuciosamente el resto de pulsos centrales y periféricos, la auscultación cardiaca, vascular y la presión arterial, para detectar posibles alteraciones vasculares. Aunque el paciente no refiera síntomas visuales o neurológicos, es importante realizar una exploración oftalmológica y neurológica exhaustiva.
Pruebas complementarias
- Analítica. Hemograma completo, coagulación y bioquímica con pruebas de función renal, perfil hepático y lipídico, ácido úrico, creatina-cinasa, ferritina, hemoglobina glicosilada, perfil tiroideo, proteinograma, inmunoglobulinas, calcio, fósforo, fosfatasa alcalina, PTH, vitamina D, estudio básico de orina, complemento, VSG, PCR, factor reumatoide, anticuerpos antipéptidos citrulinados cíclicos, ANA, ANCA y crioglobulinas (según sospecha clínica).
- Serologías
- VIH, sífilis, VHC, VHB, VHA, CMV, toxoplasma, VVZ, sarampión, rubeola y parotiditis.
- Si antiHBc y/o HBsAg positivos y VHC IgG positivos: carga viral.
- Argentina, Belice, Bolivia, Brasil, Chile, Colombia, Ecuador, El Salvador, Guatemala, Guayana, Guayana francesa, Honduras, México, Nicaragua, Panamá, Paraguay, Perú, Surinam, Uruguay y Venezuela: serología de Trypanosoma cruzi (enfermedad de Chagas).
- África: serología de Schistosoma spp. y examen en fresco de orina.
- África, Latinoamérica y Asia: serología de Strongyloides stercoralis.
- Mantoux y Quantiferón: despistaje de tuberculosis latente.
- Electrocardiograma.
- Pruebas de imagen (8, 9, 10, 11, 12, 13)
- Radiografía posteroanterior y lateral de tórax.
- Radiografía anteroposterior y lateral de columna dorsal y lumbar.
- Ecografía Doppler de arteria temporal, axilar y troncos supraaórticos (carótida común, carótida interna y externa, temporal superficial rama frontal y rama parietal, vertebral, subclavia y axilar):
- Se recomienda como primera modalidad de imagen en pacientes con sospecha de ACG (Figura 1).
- En pacientes con alta sospecha clínica de ACG y una prueba de imagen positiva el diagnóstico de ACG puede realizarse sin una prueba adicional, como la biopsia de arteria temporal u otros estudios de imagen.
- La detección de estenosis u oclusiones en las arterias temporales puede ser indicativo de arteritis temporal, pero no es específico porque puede verse en otras enfermedades como la arterioesclerosis. Por ello, no es un dato suficiente para el diagnóstico.
- Su realización puede ayudar a seleccionar el punto de la biopsia.
- Realizar ante un resultado negativo de la biopsia de arteria temporal.
- Realizar en caso de no tener acceso a la realización de la biopsia o bien se prevea que la biopsia se va a demorar en el tiempo.
- El estudio ecográfico tiene que incluir:
- Presencia o ausencia del signo del halo en arteria temporal y vertebral: engrosamiento de la pared arterial homogéneo, hipoecoico y bien delimitado, endoluminal, visible tanto en el plano longitudinal como el transversal y habitualmente concéntrico.
- Presencia o ausencia del signo de la compresión: engrosamiento de la pared arterial (halo) que permanece visible como un área hipoecoica homogénea tras aplicar presión con el transductor.
- Otras variables ecográficas que se pueden realizar:
- Velocidad pico sistólico máximo en las ramas y el tronco de la arteria temporal en cortes longitudinales.
- Velocidades y flujos en las arterias vertebrales.
- Presencia o ausencia de oclusión vascular: ausencia de señal Doppler en una arteria visible llena de material hipoecoico, incluso con baja frecuencia y alta ganancia de color.
- Signo de la órbita vacía: falta de visualización de arterias ciliares posteriores (Fig. 1). Su detección implica riesgo de afectación ocular, por lo que debe valorarse la corticoterapia urgente si se detecta.
- Grosor íntima-media (GIM): si existe signo del halo y se confirma el diagnóstico, se puede determinar el GIM de la arteria con los signos vasculíticos para el seguimiento. Esta determinación incrementa la sensibilidad y especificidad. Solo sería valorable el GIM para el seguimiento en la arteria con signo de halo al diagnóstico y siempre que se excluya que haya placas ateromatosas a ese nivel. Los puntos de corte son los siguientes: arteria temporal ≥ 0,34 mm; gran vaso ≥ 1 mm.
- En caso de no estar disponible la ecografía, la resonancia magnética de alta resolución (preferiblemente) o el FDG-PET pueden utilizarse como alternativas a la ecografía para la evaluación de las arterias craneales.
- Otras pruebas de imagen. En pacientes con sospecha de afectación de arterias extracraneales se aconseja realizar pruebas no invasivas de imagen vascular para valorar la afectación de la aorta y sus ramas principales. En tal caso, debe solicitarse con carácter preferente tomografía por emisión de positrones (PET), y como alternativa, angio-TC o angio-RM. Se debe realizar PET/TC en las siguientes situaciones:
- ACG oculta o enmascarada.
- Fenotipo con sospecha de afectación de gran vaso sin compromiso craneal.
- En los casos de alta sospecha clínica con ecografía y biopsia de arteria temporal negativas, para intentar obtener una confirmación objetiva de la presencia de vasculitis.
- Biopsia de arteria temporal unilateral (2-5 cm)
- • Es la técnica de elección.
- Permite confirmar la ACG.
- Una biopsia negativa no excluye el diagnóstico.
- Permite descartar otras causas de vasculitis (poliarteritis microscópica, Wegener, poliarteritis nodosa, crioglobulinemia, Buerguer, vasculitis reumatoide y lúpica) y enfermedad no vasculítica (amiloidosis primaria, linfoma, sarcoidosis, calcifilaxis, arteriosclerosis, herpes zóster, endocarditis, fiebre Q y sífilis).
- Se debe realizar tan pronto como sea posible, preferiblemente antes de las 2 semanas.
- La biopsia deberá llevarse a cabo en unidades de cirugía con experiencia en su realización.
- Anatomía patológica: fragmentación de la elástica interna de la pared vascular junto con la presencia de infiltrado inflamatorio con predominio de células mononucleares o inflamación granulomatosa, frecuentemente células gigantes multinucleadas.
- En función de la localización del infiltrado inflamatorio se han descrito 4 patrones histológicos:
- Inflamación transmural (más frecuente).
- Inflamación en la zona de unión entre la íntima y la media o en la adventicia.
- Inflamación en los vasa vasorum intramurales (vasa vasorum vasculitis).
- Inflamación en los pequeños vasos adventiciales localizados alrededor de una arteria temporal normal (small vessel vasculitis).
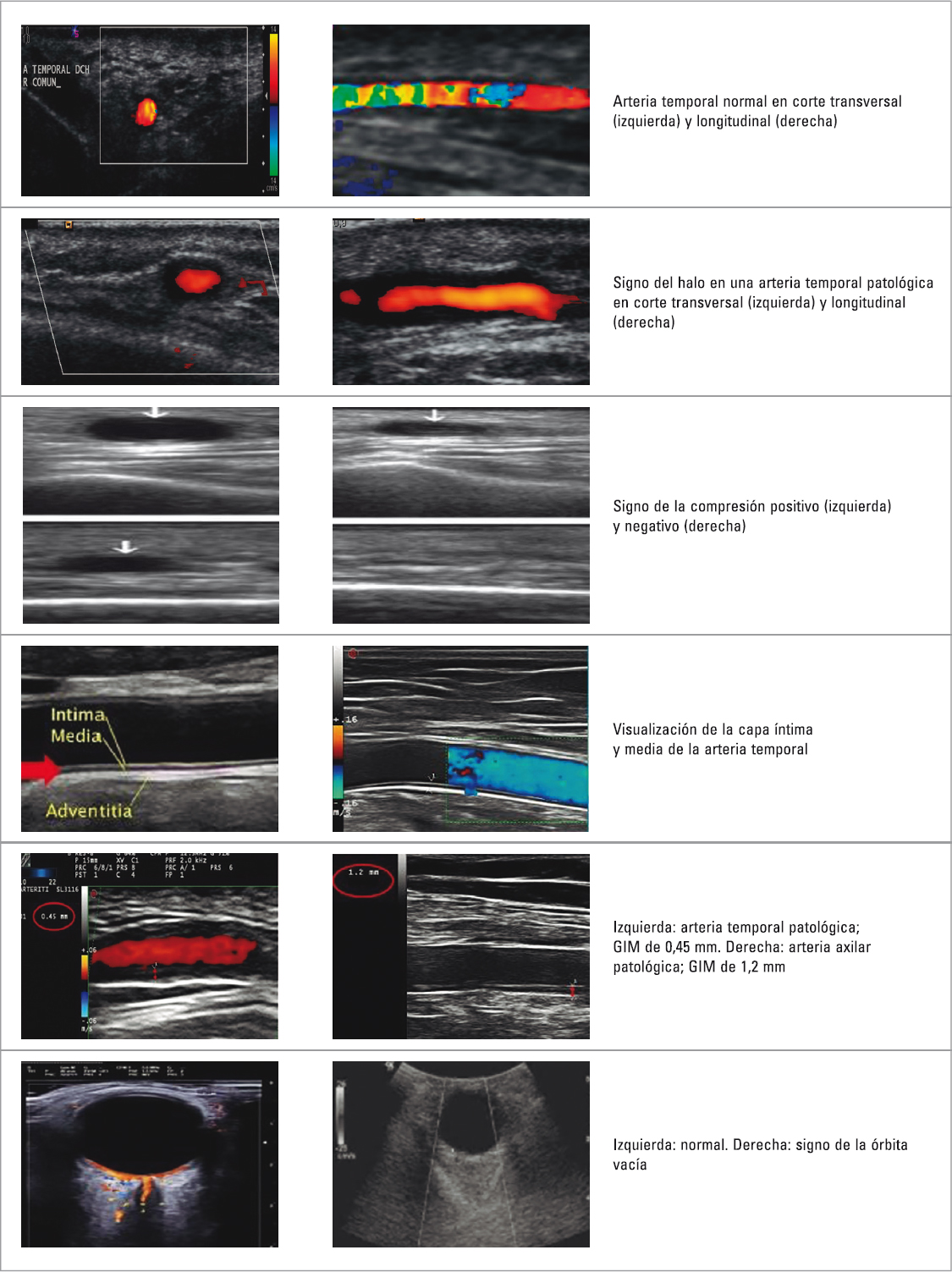
FIGURA 1. Imágenes ecográficas obtenidas del Fondo de Imágenes de la Sociedad Española de Reumatología (autor: Dr. Eugenio de Miguel Mendieta) y cedidas por el Servicio de Neurología del Hospital Universitario Virgen Macarena, Sevilla. Disponible en: https://fondodeimagen.ser.es. GIM: grosor íntima-media.
Manejo
Los pacientes con sospecha de ACG se benefician de un abordaje multidisciplinar para afrontar el diagnóstico de la enfermedad. Se deberá garantizar la aplicación de protocolos consensuados en colaboración con los Servicios de Anatomía Patología, Urgencias, Radiología, Oftalmología, Neurología, Reumatología o Medicina Interna.
Con independencia de la gravedad del cuadro clínico, la ACG es una urgencia médica y el inicio del tratamiento no debe demorarse14,15.
Tratamiento de la enfermedad grave
En caso de vasculitis con manifestaciones que amenazan la vida o los órganos (ictus, afectación ocular, necrosis facial, necrosis lingual, enfermedad cardiaca, afectación de aorta y sus ramas, isquemia de extremidades) el paciente debe quedar ingresado en planta de hospitalización. El abordaje terapéutico es el siguiente:
- Pulsos de metilprednisolona i.v. (250-1000 mg/día) durante 3 días en función de la situación clínica del paciente.
- Tras los pulsos, glucocorticoides orales (prednisona o equivalente v.o, preferiblemente en dosis única matinal, pudiendo en casos seleccionados optar por dosis fraccionada) a dosis de 1 mg/kg/día (máximo 60 mg/día) en pauta descendente (Tabla 1, izquierda).
- AAS 100 mg/día de forma continuada en caso de estenosis significativa en territorio de los troncos supraaórticos o intracraneales.
- Omeprazol 20 mg/día de forma continuada.
- Tratamiento preventivo de la osteoporosis asociada a corticoides: calcio, vitamina D y fármacos antirresortivos (seguir las guías de osteoporosis).
- Profilaxis de Pneumocystis Jirovecci: trimetoprima/sufametoxazol (Septrin®) 160 mg/800 mg en días alternos (3 días en semana) o dosis de 80 mg/ 400 mg diarios. En caso de intolerancia o alergia se puede sustituir por pentamidina inhalada (300 mg mensuales) o atovacuona 1,5 mg/día. Suplementar con ácido fólico si se prevé que el tratamiento va a durar más de un mes. Una vez se suspendan los glucocorticoides o se mantengan a dosis bajas, valorar suspender la profilaxis.
- Profilaxis de tuberculosis latente en los casos indicados.
- Control de los factores de riesgo cardiovascular.
- Fármacos inmunosupresores. En la enfermedad grave se aconseja el uso concomitante de glucocorticoides más tocilizumab a dosis de 162 mg semanal por vía subcutánea o de 4-8 mg/Kg/mes por vía intravenosa. La decisión de tratar con tocilizumab más glucocorticoides, metotrexato más glucocorticoides o glucocorticoides en monoterapia para la terapia inicial debe basarse en el estado clínico del paciente. Si bien existe una mayor evidencia que respalda el uso de tocilizumab en comparación con metotrexato, se puede considerar metotrexato en pacientes que no pueden usar tocilizumab debido a factores tales como como infecciones recurrentes, antecedentes de perforaciones gastrointestinales o diverticulitis.
- Si se produce un empeoramiento o persisten las manifestaciones isquémicas, plantear revascularización (p.ej. angioplastia). Valorar en estos casos anticoagulación o doble antiagregación. En la medida de lo posible, las intervenciones endovasculares electivas o la cirugía reconstructiva deben realizarse en fases de estabilidad o remisión. Sin embargo, la disección de vasos arteriales y la isquemia vascular crítica requieren una derivación urgente a un equipo vascular. A nivel neurológico (estenosis de troncos supraaórticos o intracraneales) no existe evidencia de la anticoagulación ni la revascularización en las estenosis vasculíticas. El seguimiento de las complicaciones neurológicas será realizado por la unidad de Neurología correspondiente.
TABLA 1. Pautas de prednisona recomendadas en la arteritis de células gigantes
| 109 semanas | 26 semanas | ||
|---|---|---|---|
| Dosis | Duración | Dosis | Duración |
| 1 mg/kg/día (máx. 60 mg/día) | 4 semanas | 60 mg/día | 1 semana |
| 50 mg/día | 1 semana | 50 mg/día | 1 semana |
| 40 mg/día | 1 semana | 40 mg/día | 1 semana |
| 30 mg/día | 1 semana | 35 mg/día | 1 semana |
| 20 mg/día | 2 semana | 30 mg/día | 1 semana |
| 15 mg/día | 4 semanas | 25 mg/día | 1 semana |
| 10 mg/día* | 3 meses | 20 mg/día | 1 semana |
| 10/7,5 mg/día (días alternos) | 3 meses | 15 mg/día | 1 semanas |
| 7,5 mg/día | 3 meses | 12,5 mg/día | 2 semanas |
| 7,5/5 mg/día (días alternos) | 3 meses | 10 mg/día | 2 semanas |
| 5 mg/día | 3 meses | 7,5 mg/día | 3 semanas |
| 5/2,5 mg/día (días alternos) | 3 meses | 5 mg/día | 3 semanas |
| 2,5 mg/día | 3 meses | 2,5 mg/día | 4 semanas |
| 2,5/0 mg/día (días alternos) | 3 meses y suspender | 2,5 mg/48 horas | 4 semanas y suspender |
|
* A partir de 10 mg/día, la duración del mantenimiento y la posterior reducción es orientativa. Es aconsejable que la reducción siguiente se inicie un mes antes de la visita de control, con ello se podrá observar la tolerancia a la reducción. |
|||
Tratamiento de la enfermedad no grave
En caso de vasculitis sin manifestaciones que amenacen la vida o los órganos (síntomas constitucionales, cefalea, hipersensibilidad en cuero cabelludo, claudicación mandibular, polimialgia reumática, artritis, tenosinovitis) se recomienda el mismo esquema de tratamiento que en la enfermedad grave, salvo que no están indicados los pulsos de metilprednisolona.
- Glucocorticoides orales (prednisona o equivalente v.o, preferiblemente en dosis única matinal, pudiendo en casos seleccionados optar por dosis fraccionada) a dosis de 1 mg/kg/día (máximo 60 mg/día) en pauta descendente (Tabla 1, derecha).
- Se puede realizar un seguimiento estrecho en consulta sin necesidad de ingreso con solicitud de pruebas complementarias de carácter preferente e inicio de tratamiento inmediato.
- Se valorará la necesidad de uso concomitante de glucocorticoides más terapia inmunosupresora según la situación clínica del paciente (vide infra indicaciones de tratamiento con tocilizumab).
Consultas sucesivas
Inicialmente, se realizarán controles frecuentes mensuales o bimensuales según la clínica, con posteriores visitas cada 3-4 meses hasta completar el primer año de tratamiento. Si transcurrido ese periodo se ha conseguido una estabilidad tanto clínica como analítica y no se han producido rebrotes, en el segundo año se pueden realizar controles cada 4-6 meses. Se recomienda en cada visita:
- Anamnesis detallada y exploración física.
- Analítica con hemograma, VSG, PCR, función renal y hepática, hemoglobina glicosilada y perfil lipídico. La actividad de la enfermedad se monitoriza mejor con la PCR, más sensible que la VSG.
- Control estricto y tratamiento de los factores de riesgo cardiovascular, como la glucemia, la tensión arterial y la dislipemia.
- Control de la iatrogenia inducida por el tratamiento corticoideo y otros inmunosupresores.
Tras el primer año de tratamiento corticoideo se realizará una interconsulta a Oftalmología para revisión oftalmológica que incluya examen de la agudeza visual y fondo de ojo de control en pacientes con alteraciones oculares en el inicio de la enfermedad.
Se recomienda el despistaje de aneurismas aórticos de forma periódica, cada 2-4 años, mediante angio-TC o angio-RM. Entre los 3 y los 10 años tras el diagnóstico se sitúa el mayor riesgo de desarrollo de aneurismas aórticos, por lo que se recomienda su despistaje, incluso sin la administración de contraste. El TAC toraco-abdominal tiene una excelente resolución espacial para medir con exactitud el diámetro del vaso. Una vez diagnosticados, se debe repetir anualmente para valorar la progresión de la dilatación, y se contará con la valoración y el control por la unidad de Cirugía Vascular.
Tratamiento según evolución
Mejoría clínica. Reducir progresivamente la dosis de glucocorticoides hasta su suspensión si se produce remisión clínica. La duración aproximada del tratamiento suele ser 2-3 años (Tabla 1, izquierda), sin embargo, hasta el 50% requieren pautas más prolongadas con dosis de mantenimiento inferior a 10 mg/día. En pacientes con terapia inmunosupresora como tocilizumab, si el paciente presenta mejoría clínica, los glucocorticoides se pueden retirar antes, aproximadamente a las 26 semanas (Tabla 1, derecha). Se debe mantener dosis de tocilizumab al menos un año en pauta semanal. Posteriormente, si hay buena evolución, se podrá espaciar a cada 2 semanas, valorando su retirada si el paciente se encuentra en remisión clínica de forma mantenida.
Remisión. Ausencia de todos los signos y síntomas clínicos atribuibles a ACG activa con normalización de VSG y PCR. En pacientes con enfermedad extracraneal no debe haber evidencia de dilatación o estrechamiento progresivo de los vasos.
Recaída. Reaparición de los síntomas y aumento significativo de los reactantes de fase agua, con o sin aparición de anemia, una vez descartadas otras causas.
- Recaída menor. Recurrencia de la enfermedad activa que no cumple los criterios de recaída mayor.
- Aumentar la dosis de prednisona 5-10 mg (dependiendo de la intensidad) por encima de la dosis que controlaba el cuadro clínico, mantener durante 4 semanas y continuar con el esquema terapéutico habitual.
- Recaída mayor. Recurrencia de la enfermedad activa con cualquiera de los siguientes: características clínicas de isquemia (incluida la claudicación mandibular, síntomas visuales, pérdida visual atribuible a ACG, necrosis de cuero cabelludo, ictus, claudicación de extremidades) y/o evidencia de inflamación aórtica activa que resulta en dilatación progresiva de vasos grandes o aórticos, estenosis o disección.
- Valorar pulsos de metilprednisolona.
- Aumentar la dosis de glucocorticoides a 40-60 mg/día.
- Valorar añadir un ahorrador de glucocorticoides (tocilizumab o metotrexato).
En caso de rebrotes frecuentes o en pacientes que hayan desarrollado o tengan un alto riesgo de desarrollar efectos adversos de los glucocorticoides, como osteoporosis, diabetes o glaucoma, se debe plantear tratamiento adyuvante con metotrexato o tocilizumab como alternativa.
- Metotrexato: 10-25 mg semanales vía oral o subcutánea, siempre con ácido fólico 5 mg semanales el día siguiente del metotrexato.
- Tocilizumab: 162 mg por vía subcutánea semanal o dosis de 4-8 mg/kg/mes por vía intravenosa.
El aumento aislado de la VSG sin un cuadro clínico acompañante no debe considerarse rebrote de la enfermedad. En este caso se aconseja descartar otras causas y mantener el mismo tratamiento, junto con un control más estrecho y visitas más frecuentes.
Enfermedad refractaria. Incapacidad de alcanzar la remisión a pesar del tratamiento estándar recomendado (normalmente, que requiera dosis de prednisona ≥ 10 mg/día para su control). Debe valorarse terapia adyuvante con tocilizumab o metotrexato. Las indicaciones y recomendaciones para iniciar el tratamiento con tocilizumab se listan a continuación:
- Pacientes con enfermedad grave, como síntomas oculares, isquemia cerebral o afectación de grandes vasos extracraneales.
- Pacientes con enfermedad no grave que experimentan una recaída de la enfermedad con síntomas graves mientras reciben glucocorticoides.
- Pacientes con enfermedad no grave que experimentan una recaída de la enfermedad mientras reciben glucocorticoides en dosis moderadas a altas.
- Pacientes con enfermedad no grave refractaria al tratamiento con glucocorticoides.
- Comorbilidades que contraindique una pauta larga de glucocorticoides: diabetes mellitus, hipertensión arterial, osteoporosis, hemorragia gastrointestinal, glaucoma y cataratas.
En pacientes que no puedan usar tocilizumab debido a factores tales como como infecciones recurrentes, antecedentes de perforación gastrointestinal o diverticulitis, considerar el uso metotrexato.
MODELO DE ACTUACIÓN DIAGNÓSTICA Y DE TRATAMIENTO A REALIZAR EN URGENCIAS
La figura 2 expone el modelo de actuación propuesto en el Servicio de Urgencias del Hospital Universitario Virgen Macarena (Sevilla) ante un paciente con sospecha de ACG. En caso de sospecha de ACG sin que cumpla criterios diagnósticos se recomienda ingresar al paciente para su reevaluación.
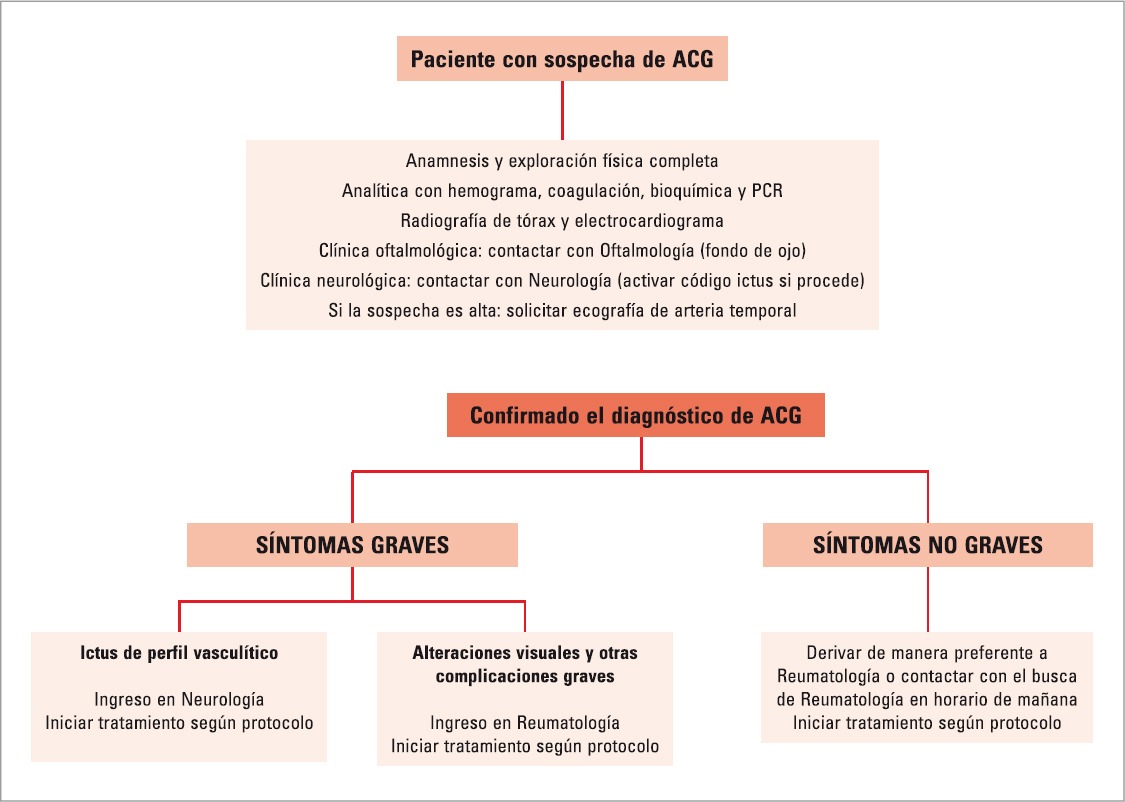
FIGURA 2. Protocolo de actuación en el Servicio de Urgencias del Hospital Universitario Virgen Macarena, Sevilla. ACG: arteritis de células gigantes; PCR: proteína C reactiva.
FINANCIACIÓN
El presente trabajo no ha recibido ninguna subvención oficial, beca o apoyo de un programa de investigación destinados a la redacción de su contenido.
CONFLICTO DE INTERESES
Los autores no comunican conflicto de intereses en relación con el contenido del trabajo.
RESPONSABILIDADES ÉTICAS
Protección de personas y animales
Los autores declaran que para este trabajo no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos
Los autores declaran que en este trabajo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado
Los autores declaran que en este trabajo no aparecen datos de pacientes.
Uso de inteligencia artificial generativa
Los autores declaran que no han utilizado ningún tipo de inteligencia artificial generativa en la redacción de este manuscrito ni en la creación de figuras, gráficos, tablas o sus correspondientes pies o leyendas.
