Introducción
Las causas de los trastornos neurológicos son diversas e incluyen infecciones, traumas, factores ambientales, factores inmunitarios, etc. Adicionalmente, en los últimos años, la evidencia ha puesto de manifiesto la importante contribución de los factores genéticos en el desarrollo y progresión de numerosas enfermedades neurológicas.
En función de la contribución de la genética en el desarrollo de una enfermedad, podemos clasificar las enfermedades en complejas y monogénicas1. La mayoría de las enfermedades comunes son complejas y su causa es multifactorial, es decir, su desarrollo es dependiente de la interacción de múltiples factores tanto genéticos como ambientales y de estilo de vida. En las enfermedades monogénicas, también conocidas como mendelianas por seguir los patrones de herencia descritos por Mendel, variantes genéticas en un solo gen son suficientes para causar enfermedad. No obstante, entre las formas complejas y monogénicas existe en realidad un espectro continuo de contribución genética (Fig. 1).
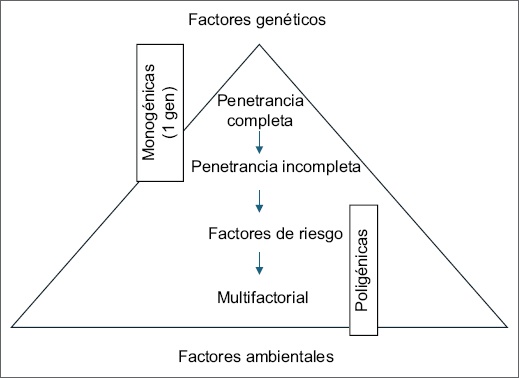
Figura 1. En el primer nivel tenemos las formas monogénicas de enfermedad con penetrancia completa, en las que, de modo general, un individuo que presente una variante patogénica en un gen (o dos variantes patogénicas en el caso de herencia recesiva) va a desarrollar la enfermedad. En el segundo nivel tendríamos las formas monogénicas con penetrancia incompleta, en las que solo algunos de los individuos que presentan la variante van a desarrollar enfermedad; la expresión clínica depende de otros factores modificadores. En el siguiente nivel tendríamos los factores de riesgo, que incrementan el riesgo de desarrollar enfermedad de forma significativa, aunque no son suficientes por sí solos para causarla. En el extremo final tendríamos la causa multifactorial, en la que el desarrollo de la enfermedad depende de la interacción de múltiples factores genéticos y ambientales, en este escenario ningún factor es determinante por sí solo.
En algunas enfermedades neurológicas, como la enfermedad de Parkinson, la mayoría de los casos presentan un origen multifactorial, mientras que un pequeño porcentaje de pacientes presenta formas mendelianas asociadas a variantes patogénicas en genes específicos.
Actualmente, en la práctica clínica, los estudios genéticos se dirigen principalmente a las formas monogénicas de enfermedad. En este contexto, identificar la causa genética tiene utilidad clínica a varios niveles: permite establecer o confirmar el diagnóstico, proporcionar un asesoramiento genético y reproductivo más preciso al paciente y a sus familiares, mejorar la estimación del pronóstico, orientar el seguimiento clínico e incluso evita la realización exploraciones o procedimientos innecesarios. Asimismo, en determinados casos, la identificación de la alteración genética puede guiar o determinar el tratamiento del paciente. Cabe destacar que, en los últimos años, el número de terapias dirigidas frente a la causa molecular ha aumentado de forma notable; en estos casos, la identificación de la variante genética resulta esencial2.
En el marco de la etiología multifactorial se han identificado numerosas variantes de susceptibilidad o factores de riesgo, principalmente mediante estudios de asociación del genoma completo (GWAS, genome-wide association studies). Entre las herramientas emergentes para integrar el efecto conjunto de estas variantes destacan las puntuaciones de riesgo poligénico (PRS, polygenic risk score). Estas combinan el efecto acumulativo de numerosas variantes genéticas comunes para estimar la predisposición genética individual a desarrollar una determinada enfermedad o rasgo. El cálculo del PRS se basa en la suma ponderada de los alelos de riesgo presentes en un individuo, donde el peso de cada variante se ha otorgado en base al tamaño de su efecto estimado en estudios previos de GWAS3. Aunque su implementación en el campo de la neurología todavía es limitada, en otros campos se utilizan algoritmos que integran los PRS en combinación con factores no genéticos (p. ej., sexo, edad, estilo de vida, etc.) que permiten estratificar a los individuos según su riesgo genético demostrando su potencial como herramienta complementaria en estrategias de prevención, cribado o manejo personalizado4,5.
Tipos estudios genéticos
Los estudios genéticos pueden clasificarse según el tipo de variantes que permiten detectar y la extensión del análisis realizado.
La secuenciación Sanger permite identificar variantes de secuencia, como las de nucleótido único (SNV, single nucleotide variants) y pequeñas inserciones y deleciones (indels, contracción de «inserción o deleción») (Fig. 2) en regiones de interés de hasta aproximadamente 1.000 pares de bases (pb). Con el desarrollo de las tecnologías de secuenciación de nueva generación (NGS, next generation sequencing), que permiten secuenciar millones de fragmentos de ADN de forma masiva y en paralelo, ha sido posible ampliar el alcance del análisis genético desde la secuenciación de unos pocos genes hasta la del genoma completo. En la actualidad, la secuenciación Sanger ha sido en gran medida reemplazada por las tecnologías NGS, y su uso se limita principalmente a los estudios de segregación familiar o a la confirmación de variantes en regiones técnicamente complejas.
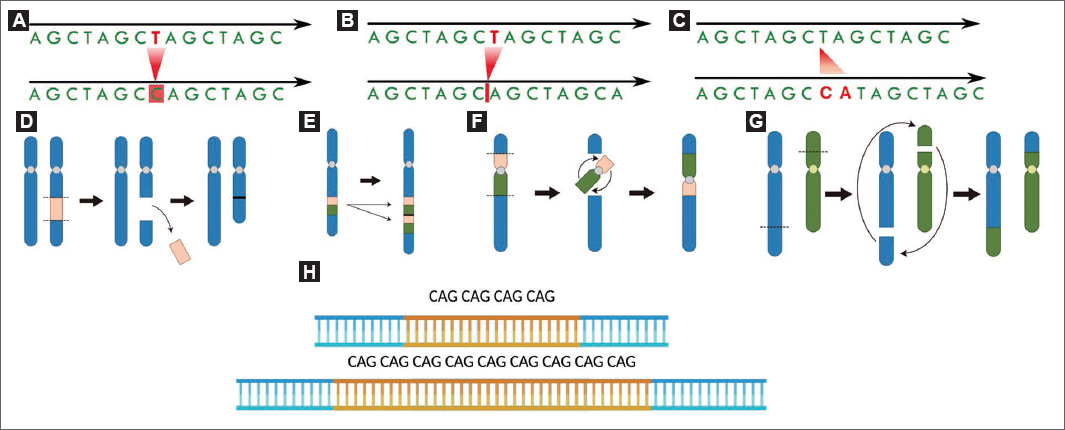
Figura 2. Clasificación esquemática de las variantes genéticas según su tamaño y naturaleza (imágenes adaptadas de Bioicons [DOI: 10.5281/zenodo.11068293] y BioRender). Panel superior: variantes de secuencia (≤ 50 pb). A: variantes de nucleótido único (SNV). B: deleciones tipo indel. C: inserciones tipo indel. Panel medio: variantes estructurales (> 50 pb). Deleciones (D), duplicaciones (E), traslocaciones (F) e inversiones (G). Panel inferior: expansiones de repeticiones en tándem (H).
En el ámbito de las tecnologías de NGS, en función del alcance de la secuenciación pueden distinguirse principalmente tres estrategias: secuenciación por librería o dirigida, secuenciación del exoma completo (WES, whole exome sequencing) y secuenciación del genoma completo (WGS, whole genome sequencing). En el caso de las librerías, se realiza un enriquecimiento específico para capturar regiones de interés del genoma, generalmente un conjunto de genes asociados a un determinado grupo de afecciones. Por su parte, el WES permite secuenciar las regiones codificantes del genoma, que representan aproximadamente el 2% de este y que se estima que contienen el 85% de las variantes causantes de enfermedad conocidas6. Finalmente, el WGS permite secuenciar la totalidad del genoma, incluidas las regiones no codificantes, como las regiones intrónicas, promotoras y reguladoras.
El análisis de los datos de secuenciación, obtenidos mediante librería, WES o WGS, puede restringirse a un conjunto de genes de interés mediante el uso de paneles virtuales, lo que permite orientar el estudio hacia genes relacionados con la sospecha clínica del paciente, focalizar el análisis y reducir la identificación de hallazgos incidentales. En fenotipos complejos o con elevada heterogeneidad genética puede plantearse un abordaje más amplio orientado por fenotipo. En este tipo de análisis es habitual el uso de códigos HPO (human phenotype ontology), que proporcionan una ontología estructurada de fenotipos clínicamente relevantes, actualmente con más de 18.000 términos descritos, y permiten priorizar el análisis de variantes en genes previamente asociados con el perfil fenotípico del paciente7. También puede realizarse el análisis de varios miembros de la familia en paralelo, lo que facilita la priorización de variantes en función de su patrón de herencia. El diseño más utilizado es el análisis en trío mediante WES o WGS, en el que se estudia el caso índice junto con sus progenitores, habitualmente asintomáticos, lo que permite identificar variantes ex novo y variantes heredadas compatibles con un modelo de herencia recesiva. Se ha demostrado un incremento del rendimiento diagnóstico mediante esta aproximación, particularmente en trastornos del neurodesarrollo8.
Las variantes estructurales (SV, structural variants) comprenden alteraciones genómicas de mayor tamaño que incluyen las variantes en el número de copias (CNV, copy number variants), caracterizadas por ganancias (duplicaciones) o pérdidas (deleciones) de material genético, así como las inversiones, las translocaciones o los reordenamientos complejos (Fig. 2). Los algoritmos bioinformáticos aplicados en el WES generalmente permiten la detección de CNV, aunque con limitaciones en sensibilidad y resolución, especialmente para las de pequeño tamaño o localizadas fuera de las regiones codificantes. El WGS ofrece un mejor rendimiento para la identificación de CNV y otras SV9. No obstante, cabe mencionar que tanto el WES como el WGS, utilizados habitualmente en la práctica clínica, se basan en tecnologías de secuenciación de lectura corta (~150 pb), lo que limita la detección de SV complejas. En este contexto, las tecnologías de secuenciación de lectura larga (LRS, long-read sequencing) mejoran de forma significativa su detección, así como la de variantes en regiones genómicas complejas, tales como duplicaciones segmentarias, en genes con elevada homología con otras regiones del genoma y el estudio de expansiones de repeticiones en tándem, entre otras10.
Otra técnica ampliamente utilizada son los arrays genómicos (array-CGH/SNP arrays), que permiten el análisis de CNV a lo largo de todo el genoma. Los arrays-CGH han constituido tradicionalmente la prueba de primera línea en el estudio de los trastornos del neurodesarrollo; sin embargo, las guías más recientes recomiendan priorizar la secuenciación del exoma como prueba diagnóstica inicial en estos pacientes11. Otra técnica ampliamente utilizada para la detección de CNV es el MLPA (multiplex ligation-dependent probe amplification), que es dirigida y que permite identificar duplicaciones y deleciones, generalmente a nivel de exón, mediante sondas diseñadas frente a genes de interés.
Para el abordaje de la detección de expansiones de repeticiones en tándem (Fig. 2), causantes de enfermedad cuando superan un determinado umbral, se emplea habitualmente el análisis de fragmentos junto con la RP-PCR (repeat primer PCR)12. En la actualidad, las expansiones también pueden analizarse mediante tecnologías de LRS, que presentan ventajas frente a las técnicas convencionales, ya que permiten determinar la longitud de expansiones de gran tamaño, analizar simultáneamente múltiples motivos de repetición en distintos genes y caracterizar la composición de la secuencia, incluyendo la presencia de interrupciones intercaladas en las estructuras repetitivas13,14.
El estudio mediante WGS de lectura corta también permite la detección de expansiones si se incorporan en el pipeline programas específicos como ExpansionHunter, HipSTR o GangSTR15. No obstante, se debe tener en cuenta que su análisis tiene limitaciones, particularmente en aquellas expansiones más largas como la asociada a la ataxia espinocerebelosa 27B (SCA27B) o en expansiones comprendidas por motivos patogénicos repetitivos intercalados dentro de motivos no patogénicos como en la SCA31 o la SCA3716,17 y, en general, se requiere una validación mediante un método alternativo18.
Existe también un importante número de enfermedades neurológicas causadas por variantes patogénicas en el ADN mitocondrial (ADNmt)19. Estas variantes pueden encontrarse en proporciones variables dentro de la célula; es decir, solo un porcentaje de las copias del ADNmt contiene la variante (heteroplasmia). Este suele asociarse con enfermedad cuando supera un determinado umbral de carga mutacional. Su estudio se suele realizar mediante la secuenciación del genoma mitocondrial con NGS de alta profundidad (≈1.000X, es decir, en promedio cada posición del genoma se secuencia unas 1.000 veces), lo que permite detectar de forma precisa variantes presentes en bajo grado heteroplasmia20. Si bien algunos estudios de WES incluyen la captura del ADNmt de forma complementaria, la profundidad de lectura suele ser limitada, lo que puede dificultar la detección de variantes con bajo grado de heteroplasmia. También hay que considerar que algunas variantes pueden no detectarse en sangre y debe considerarse el estudio genético en otros tejidos (p. ej., orina, músculo o piel)21.
En la tabla 1 se pueden encontrar las técnicas principales empleadas en el estudio genético de trastornos neurológicos, con ejemplos de usos, sus diferencias y sus limitaciones.
Tabla 1. Técnicas empleadas en el estudio genético de trastornos neurológicos
| Técnica | Variantes detectables | Alcance del estudio | Indicación clínica | Ejemplos | Limitaciones |
|---|---|---|---|---|---|
| Sanger | SNV, pequeños indels (< 50 pb) | Regiones pequeñas (< 1.000 pb) | Confirmación de variantes o estudios familiares | Variantes familiares conocidas (PSEN1 en EA) | Baja escalabilidad No detecta CNV ni mosaicos de bajo grado |
| MLPA | CNV (deleciones y duplicaciones) | Genes concretos con sondas específicas | Sospecha dirigida de CNV en genes concretos | PMP22 (CMT1A/HNPP), SMN1 (AME) | No detecta SNV Estudio dirigido |
| PCR/RP-PCR | Expansión de repeticiones en tándem | Motivos de loci concretos | Sospecha clínica dirigida de la enfermedad por expansión | HTT (Huntington), FXN (ataxia de Friedreich) | Solo para loci concretos No se puede determinar con precisión la longitud de alelos expandido de gran tamaño, ni interrupciones |
| Arrays genómicos | CNV (microdeleciones y duplicaciones) | Genoma completo (resolución de 10-200 kb) | Trastornos del neurodesarrollo | Duplicación 16p11.2, Deleción 22q11.2 | No detecta SNV ni SV equilibradas Resolución dependiente de la densidad de sondas |
| Paneles NGS | SNV, indels y CNV | Grupos de genes dirigidos (típicamente de 10 a 500) | Fenotipos heterogéneos, pero bien definidos | Neuropatías, ELA | Limitado a genes contenidos en el panel |
| WES | SNV, indels y CNV | Exoma completo (~2% del genoma) | Elevada heterogeneidad genética o fenotipos inespecíficos | Epilepsias, trastornos del neurodesarrollo | Limitado a regiones codificantes |
| WGS | SNV, indels, CNV, otras SV y TR Detección en regiones no codificantes | Genoma completo | Elevada heterogeneidad genética, fenotipos inespecíficos, estudios previos negativos o sospecha de variante intrónica profunda | Fenotipos complejos | Interpretación compleja |
| Long-read sequencing | SNV, indels, CNV, SV complejas, TR, metilación, fase Detección en regiones complejas | Genoma completo o regiones específicas (lecturas > 10 kb) | Elevada heterogeneidad genética, fenotipos inespecíficos, estudios previos negativos o sospecha de variante intrónica profunda o en región compleja | Fenotipos complejos Enfermedades por expansiones |
Interpretación compleja Mayor coste (WGS) |
|
CNV: variantes en el número de copias, copy number variants; EA: enfermedad de Alzheimer; ELA: esclerosis lateral amiotrófica; kb: kilobases; pb: pares de bases; MLPA: multiplex ligation-dependent probe amplification; PCR: reacción en cadena de la polimerasa; RP-PCR: repeat primer PCR; SNV: variantes de secuencia de nucleótido único, single nucleotide variants; SV: variantes estructurales, structural variants; TR: repeticiones en tándem; WES: secuenciación del exoma completo, whole exome sequencing; WGS: secuenciación del genoma completo, whole genome sequencing. |
|||||
A continuación se presentan las principales indicaciones clínicas para la realización de estudios genéticos, organizadas según los principales grupos de trastornos neurológicos de base genética y basadas en guías clínicas y opiniones de expertos. De forma general, cabe mencionar que la ausencia de antecedentes familiares no descarta que la causa sea monogénica y, por tanto, no debe considerarse el único motivo para no indicar un estudio genético, dado que la variante puede ser de novo (aparecer por primera vez en un individuo) o seguir un patrón de herencia difícil de identificar en la historia familiar (p. ej., herencia autosómica dominante con penetrancia incompleta, herencia autosómica recesiva, etc.).
Ataxias y paraparesias espásticas
La causa de las ataxias cerebelosas es altamente heterogénea e incluye tanto múltiples causas adquiridas como genéticas. En pacientes con ataxia de inicio tardío deben descartarse inicialmente las causas adquiridas, especialmente en aquellos pacientes con historia sugestiva, inicio agudo o subagudo. Una vez excluidas, se recomienda valorar la realización de un estudio genético22–24.
Las ataxias hereditarias de presentación en edad adulta más prevalentes están causadas por la expansión de repeticiones en tándem en distintos genes (p. ej., SCA 2, 3, 6 y 27B y ataxia de Friedreich). Cabe destacar la SCA27B, identificada recientemente, que es una de las causas principales de casos de ataxia de inicio tardío en población europea25; y el síndrome de CANVAS (ataxia cerebelosa, neuropatía y arreflexia vestibular), principal causa de ataxia de inicio tardío con neuropatía26. También hay un gran número de genes en los que se han descrito variantes de secuencia y CNV26–28.
En los pacientes con sospecha de paraparesia espástica, una vez excluidas las causas adquiridas y valorado el diagnóstico diferencial, se recomienda valorar realizar un estudio genético. Las formas más frecuentes de paraparesia espástica hereditaria son SNV y CNV en el gen SPAST, aunque también existe una elevada heterogeneidad genética27,29.
Estrategia diagnóstica
- – Análisis de expansiones de repeticiones en tándem. Al menos de los genes: FXN (FRDA), ATXN1 (SCA1), ATXN2 (SCA2), ATXN3 (SCA3), CACNA1A (SCA6), ATXN7 (SCA7), TBP (SCA17), ATN1 (DRPLA), RFC1 (CANVAS) y FGF14 (SCA27B), así como FMR1 (FXTAS) en caso de temblor-ataxia e inicio en mayores de 50 años24.
- – En caso de resultado negativo en el análisis de expansiones se recomienda realizar panel de genes o exoma/genoma24.
Ante la presencia de historia familiar o clínica específica el estudio podría orientarse a un gen o grupo de genes determinados. El rendimiento diagnóstico para pacientes con ataxia es variable, depende de la estrategia y la población de estudio.
En paraparesia espástica: panel de genes o exoma/genoma29.
Miopatías
Las enfermedades musculares presentan una elevada heterogeneidad genética, habiéndose descrito más de 200 genes causales implicados en su origen. En general está recomendado el estudio de cualquier paciente con sospecha de miopatía no filiada, independientemente de la presencia de antecedentes familiares23.
Estrategia diagnóstica
- – Panel de genes o exoma/genoma.
El rendimiento diagnóstico es variable (30-50%) en función del fenotipo23. El estudio de secuenciación de ARN en músculo como prueba complementaria ha demostrado utilidad en el diagnóstico de enfermedades musculares30.
Consideraciones particulares
Ante la sospecha dirigida de las siguientes entidades se recomienda valorar si es necesaria la realización de estudios adicionales, ya que su detección por NGS puede ser limitada:
- – Sospecha de atrofia muscular espinal (AME): estudio del número de copias de SMN1/SMN2 (MLPA). La pérdida bialélica de SMN1 explica el 95% de los pacientes con AME. Para individuos con alta sospecha clínica de AME portadores de una deleción en un solo alelo del gen SMN1, se recomienda completar el estudio para descartar variantes de secuencia mediante LRS31 u otras técnicas que garanticen especificidad.
- – Sospecha de distrofia miotónica tipo 1 o tipo 2: estudio de expansión de los genes DMPK y CNBP respectivamente.
- – Sospecha de distrofia muscular oculofaríngea: estudio de la expansión del gen PABPN1.
- – Sospecha de miopatía oculofaríngea distal: estudio de expansiones de los genes ABCD3, GIPC1, LRP12, RILPL1 y NOTCH2NLC.
- – Sospecha de distrofia muscular facioescapulohumeral: estudio del número de repeticiones D4Z4 y el haplotipo de la región cromosómica 4q35 (Southern blot, optical mapping o LRS). Estudios adicionales pueden requerir análisis de metilación o secuenciación por NGS del gen SMCHD1, causante de la distrofia muscular facioescapulohumeral tipo 232.
- – Sospecha de miopatía mitocondrial: secuenciación de ADNmt. En determinados escenarios la tasa de detección en sangre puede ser baja o inexistente. Un ejemplo es la oftalmoplejía crónica progresiva externa, donde la causa principal son deleciones del ADNmt que típicamente están restringidas al músculo esquelético33.
Neuropatías
Se han descrito más de 100 genes donde la neuropatía es la única manifestación o la más prominente. El estudio genético está indicado en pacientes con sospecha de neuropatía hereditaria, una vez descartadas causas adquiridas potenciales34,35.
Estrategia diagnóstica
- – Panel de genes o exoma/genoma.
Ante sospecha de enfermedad de Charcot-Marie-Tooth desmielinizante (CMT1), formas indeterminadas de CMT o neuropatía hereditaria con susceptibilidad a la parálisis por presión (HNPP), puede considerarse como primer abordaje genético el MLPA de la región 17p11.2-p12, que incluye el gen PMP22, dada su elevada rentabilidad diagnóstica y coste-efectividad. La duplicación de esta región explica el 60-70% de los casos de CMT135, mientras que su deleción está presente en aproximadamente el 80% de los pacientes con HNPP. En casos de HNPP con resultado negativo, se recomienda completar el estudio mediante secuenciación de PMP22, responsable del ~20% restante36. El rendimiento diagnóstico es variable y dependiente del subtipo de neuropatía.
Consideraciones particulares
En caso de resultado negativo, se recomienda valorar si es necesaria la realización de estudios específicos dirigidos, dado que su detección mediante técnicas de NGS puede ser limitada35,37:
- – Valorar variantes en regiones no codificantes del gen GJB1, que pueden no estar cubiertas por NGS (panel/exoma) asociadas a CMT desmielinizante ligada al cromosoma X (CMTX1).
- – Deficiencia de sorbitol deshidrogenasa, causada por variantes bialélicas en el gen SORD, responsable de hasta un 10% de los casos de neuropatía axonal tipo CMT (CMT2) y de neuropatía motora hereditaria distal (dHMN). Este gen presenta elevada homología con el pseudogén SORD2P. En caso de alta sospecha de esta entidad (fenotipo y/o niveles de sorbitol en suero/plasma elevados) se recomiendan estudios de LRS, ya que la detección de variantes puede verse comprometida mediante NGS lectura corta38.
- – Variantes puntuales en el gen mitocondrial MT-ATP6, que podrían explicar al menos el 1% de los casos de CMT2.
- – Dentro del diagnóstico diferencial, especialmente en síndromes complejos, se recomienda valorar el estudio de la expansión en RFC1, causante de CANVAS, así como de la expansión en NOTCH2NLC, asociada a la enfermedad neuronal con inclusiones intranucleares.
Epilepsia
La mayoría de los pacientes con formas comunes de epilepsia presentan causa multifactorial. Por ello, en general existen recomendaciones específicas para solicitar estudios genéticos en aquellos pacientes en los que existe una sospecha razonable de epilepsia de causa monogénica. En esta línea, las principales indicaciones para realizar estudios genéticos, una vez descartadas previamente causas adquiridas, son39:
- – Epilepsia grave con inicio en la infancia, particularmente las encefalopatías epilépticas y del desarrollo.
- – Epilepsia con comorbilidades, principalmente discapacidad intelectual, autismo, rasgos dismórficos o manifestaciones sistémicas.
- – Síndromes específicos con causa genética conocida: epilepsia autolimitada neonatal familiar, epilepsia autolimitada infantil familiar, epilepsia hipermotora del sueño, epilepsia del lóbulo temporal con síntomas auditivos, epilepsia familiar focal con focos variables, epilepsia genética con crisis febriles plus o epilepsia mioclónica familiar del adulto.
- – Epilepsia mioclónica progresiva u otros fenotipos progresivos.
- – Epilepsia farmacorresistente, dado que identificar una causa genética puede orientar el tratamiento.
- – En contexto de estudio prequirúrgico en pacientes con epilepsia focal no adquirida farmacorresistente, ya que la identificación de una variante genética puede aportar información valiosa sobre el pronóstico y los resultados esperados del tratamiento quirúrgico40.
- – Antecedentes familiares de epilepsia significativos. Un artículo reciente señala que estudios genéticos en pacientes con al menos un familiar de primer grado afectado, o dos familiares de segundo o tercer grado con epilepsia presenta un rendimiento diagnóstico del 20% aproximadamente41.
Estrategia diagnóstica
- – Panel de genes amplio o exoma/genoma.
El rendimiento diagnóstico es variable en función del fenotipo (del 20 a > 90% en pacientes seleccionados)39.
Consideraciones particulares
Ante la sospecha clínica de las siguientes entidades, se recomienda valorar, si es necesaria, la realización de estudios adicionales, dado que su detección mediante técnicas de NGS puede ser limitada:
- – Sospecha de epilepsia mioclónica familiar del adulto: expansión de repeticiones pentanucleotídicas en los genes STARD7, YEATS2, RAPGEF2, MARCHF6, SAMD12 y TNRC6A42.
- – Sospecha de epilepsia mioclónica progresiva: valorar, de forma adicional al estudio de NGS el estudio de la expansión en el gen CSTB, causante de la enfermedad de Unverricht-Lundborg.
- – Sospecha de síndrome de cromosoma 20 en anillo (crisis frontales refractarias, estatus epiléptico no convulsivo recurrente, tras el inicio de las crisis epilépticas deterioro cognitivo y alteraciones conductuales y EEG característico): realizar un estudio de cariotipo ampliado (estudio de mínimo 50-100 metafases)43.
- – En el contexto de la epilepsia se ha descrito de forma notable la presencia de mosaicismo somático, definido como la presencia de una variante genética en una fracción de células del individuo. En genes asociados a epilepsia y trastornos del neurodesarrollo (CDKL5, PCDH19 y SCN1A) se han descrito pacientes con variantes causales detectables en sangre periférica en mosaico44. La profundidad de cobertura de los estudios de NGS realizados, particularmente de genoma (30-50X), puede limitar la detección de mosaicismos. También cabe mencionar que, especialmente en malformaciones del desarrollo cortical, las variantes genéticas pueden estar restringidas a un subgrupo de células del tejido cerebral y no detectarse en muestras periféricas como la sangre.
Enfermedad de Alzheimer
Aproximadamente el 1% de los casos de enfermedad de Alzheimer presentan un origen mendeliano con herencia autosómica dominante, por variantes patogénicas en los genes APP, PSEN1 y PSEN2, generalmente asociadas a un inicio más precoz de la enfermedad. En general, se recomienda considerar el estudio genético en pacientes con un inicio temprano (< 60 años) o que tengan dos o más familiares con demencia. En pacientes con inicio entre 60 y 65 años o con un único familiar afectado se recomienda valorar el estudio genético en función de factores individuales (p. ej., historia familiar limitada)23,45.
El alelo e4 del gen APOE constituye el principal factor de riesgo para el desarrollo de la enfermedad de Alzheimer. Los individuos homocigotos (2% de la población) presentan un riesgo acumulado que puede alcanzar hasta el 50-60% a los 85 años, mientras que en los heterocigotos este riesgo se sitúa alrededor del 20%. Cabe mencionar que un estudio reciente sugiere una penetrancia casi completa de la biología de la enfermedad en individuos homocigotos antes de los 65 años45.
Tradicionalmente no se ha recomendado la inclusión del genotipado de APOE en el estudio genético de los pacientes con enfermedad de Alzheimer, por considerarse de valor predictivo limitado. No obstante, en los últimos años se ha reconsiderado su inclusión, especialmente tras la aprobación de tratamientos modificadores de la enfermedad como el lecanemab y el donanemab. Estos fármacos no están indicados en pacientes homocigotos para el alelo e4, debido al mayor riesgo de efectos adversos, en particular de anormalidades de imagen relacionadas con el amiloide (ARIA)45.
Estrategia diagnóstica
- – Panel de genes que incluya APP, PSEN1 y PSEN2. Considerar genotipado de APOE.
El rendimiento diagnóstico del estudio es del 5 al 13% en casos de inicio temprano46.
Demencia frontotemporal
Aproximadamente el 30% de los casos de demencia frontotemporal (DFT) presentan una causa monogénica, en su mayoría con herencia autosómica dominante. Las causas más frecuentes son la expansión en C9orf72 y variantes en los genes GRN y MAPT.
Se recomienda el estudio genético en pacientes con fenotipo clínico de DFT/esclerosis lateral amiotrófica (ELA) o variante conductual de DFT, incluso en ausencia de antecedentes familiares. En otros fenotipos de DFT, el estudio debe valorarse de forma individualizada, e indicarse especialmente en presencia de historia familiar. En las afasias primarias progresivas atípicas debe considerarse el estudio genético ante la posibilidad de variantes en el gen GRN45.
Estrategia diagnóstica
- – Estudio expansión gen C9orf72 y panel genes que incluya al menos los genes MAPT y GRN.
Consideraciones particulares
Valorar dentro del diagnóstico diferencial el estudio de la expansión del gen PRNP, causante de enfermedad priónica47.
Esclerosis lateral amiotrófica
En la mayoría de los pacientes con ELA familiar se identifican variantes genéticas asociadas a formas mendelianas (50-85% de los pacientes), así como en algunos casos de ELA esporádica (hasta el 10-20% de los pacientes sin antecedentes familiares). Actualmente, se recomienda ofrecer el estudio genético a cualquier paciente con ELA, independientemente de los antecedentes familiares48. La causa genética más común de ELA en población europea es la expansión en el gen C9orf72, seguida de variantes en el gen SOD1, para las que actualmente se dispone de una terapia dirigida (tofersén).
Estrategia diagnóstica
- – Estudio de expansión del gen C9orf72 y panel de genes que incluya al menos los genes SOD1, TARDBP y FUS1. Se recomienda además la inclusión de otros genes con evidencia sólida de asociación con ELA48.
Consideraciones particulares
- – Valorar adicionalmente el estudio de expansiones en los genes ATXN2 y LRP12, en los que la ELA se considera parte del espectro fenotípico de la SCA tipo 2 y de la miopatía oculofaríngea distal tipo 1, respectivamente, aunque la casuística disponible es limitada49,50.
- – Considerar también dentro del diagnóstico diferencial el estudio de la expansión del gen AR, causante de la atrofia muscular espinal y bulbar (o enfermedad de Kennedy) en varones51, además del MLPA de SMN1/SMN2 para el estudio de AME.
Enfermedad de Parkinson
Se estima que aproximadamente un 10% de los pacientes con enfermedad de Parkinson presentan causa mendeliana. De forma general, se recomienda el estudio genético a los pacientes con inicio temprano (< 50 años), etnias de alto riesgo (judía asquenazí y bereber africana) o antecedentes familiares de enfermedad de Parkinson u otro trastorno del movimiento o demencia52. En los últimos años, algunos expertos apuntan a que debería considerarse incorporar el estudio genético como parte de la rutina diagnóstica en la enfermedad de Parkinson52,53, considerando en parte la existencia de ensayos clínicos dirigidos a la causa molecular.
Estrategia diagnóstica
- – Panel de genes que incluya al menos los genes LRRK2, PRKN, SNCA, PINK1, PARK7 y VPS35.
Variantes en el gen GBA1 se consideran el principal factor de riesgo en la enfermedad de Parkinson. La presencia de variantes patogénicas en este gen se asocia con un mayor riesgo de desarrollar la enfermedad con un incremento del riesgo respecto a la población general de entre el 2 y 15% según la población y variante estudiada54,55. La detección de variantes en GBA1 mediante técnicas de NGS convencionales, como el exoma, puede verse comprometida debido a su elevada homología con un pseudogén cercano (GBAP1).
Distonía
La distonía es un trastorno del movimiento heterogéneo clínica y genéticamente. La probabilidad de identificar una causa monogénica es mayor en pacientes con inicio temprano (antes de los ≈20-30 años), distribución extensa (distonía segmentaria o generalizada), distonía combinada o presencia de síntomas neurológicos adicionales no relacionados con trastornos del movimiento. Un algoritmo diagnóstico que permite seleccionar a los pacientes para estudio genético en los que se estima un rendimiento diagnóstico por exoma del 25% o superior, puede encontrarse en este trabajo56.
Estrategia diagnóstica
- – Panel de genes o exoma/genoma.
Enfermedad de Huntington
La causa de la enfermedad de Huntington es una expansión del trinucleótido CAG en el exón 1 del gen HTT57.
Estrategia diagnóstica
- – Estudio de expansión del gen HTT.
Consideraciones particulares
Ante un resultado negativo se han de considerar genes causantes de fenocopias de Huntington (formas Huntington-like): estudio de la expansión del gen JPH3, causante de Huntington-like tipo 2 y fenocopia más común, particularmente en pacientes de ascendencia africana; expansión gen TBP asociada a ataxia espinocerebelosa tipo 17; expansión del gen ATN1 asociada a atrofia dentato-rubro-pálido-luisiana (DRPLA); expansión del gen PRPN causante de una prionopatía y considerada como Huntington-like tipo 1, y la expansión de C9orf72. Adicionalmente, se recomienda valorar un panel de genes o exoma/genoma, dado que se han reportado variantes de secuencia en otros genes asociadas a otras fenocopias o formas de corea58.
Ictus/patología neurovascular
La mayoría de los casos de ictus presentan una causa multifactorial; sin embargo, entre el 1-5% se deben a causas monogénicas59. Se recomienda realizar un estudio genético ante la sospecha de etiología mendeliana: ictus de inicio precoz (antes de los 55 años), la presencia de un fenotipo sugestivo de una entidad monogénica, manifestaciones sistémicas asociadas, un fenotipo clínico inusual o antecedentes familiares, especialmente cuando siguen un patrón de herencia mendeliana60,61. La presencia de múltiples factores de riesgo cardiovascular no excluye la posibilidad de un origen monogénico60,61.
Una de las causas principales de ictus es la enfermedad de pequeño vaso cerebral. Su causa monogénica más común es la arteriopatía cerebral con infartos subcorticales y leucoencefalopatía (CADASIL), originada por variantes genéticas en NOTCH361. Otras afecciones neurovasculares relevantes son la cavernomatosis múltiple familiar, causada por variantes genéticas en KRIT1, CCM2 y PDCD10, y la arteriopatía de moyamoya62. Algunas enfermedades neurovasculares hereditarias cursan con leucoencefalopatía, por lo que deben contemplarse en su estudio.
Estrategia diagnóstica
- – Panel de genes o exoma/genoma62.
Consideraciones particulares
Considerar dentro del diagnóstico diferencial el síndrome de MELAS (encefalopatía mitocondrial, acidosis láctica y episodios stroke-like) causado por variantes genéticas en el ADNmt, la mayoría de los casos causados por la variante recurrente m.3243A>G en MT-TL163.
Leucoencefalopatías/leucodistrofias
Las leucoencefalopatías y leucodistrofias constituyen un conjunto heterogéneo de enfermedades neurodegenerativas que afectan a la sustancia blanca del SNC y pueden deberse tanto a causas adquiridas, algunas potencialmente tratables, como a numerosos trastornos genéticos poco frecuentes, cuyo diagnóstico suele ser complejo debido a la amplia variabilidad y superposición fenotípica. Se recomienda el estudio genético en pacientes con leucoencefalopatía en los que se hayan descartado causas adquiridas, particularmente para aquellos con un patrón simétrico y confluente. En pacientes mayores de 60 años, con factores de riesgo cardiovascular significativos, evolución clínica lenta o asintomática, cambios extensos en ganglios basales y pons con presencia de lagunas crónicas y microhemorragias cerebrales se debe sospechar de enfermedad de pequeño vaso adquirida grave64.
Estrategia diagnóstica
- – Panel de genes amplio o exoma/genoma. Pruebas complementarias como estudios metabólicos pueden orientar el diagnóstico y permitir un estudio dirigido64.
Consideraciones particulares
En pacientes con sospecha de síndrome de Labrune, caracterizado por la presencia de leucoencefalopatía con quistes y calcificaciones cerebrales, debe valorarse si se ha analizado el gen SNORD118, causante de este, dado que puede no estar incluido en algunos estudios de exoma.
Interpretación de estudios genéticos
Una vez realizado el estudio genético, la interpretación de las variantes identificadas requiere de su clasificación conforme a guías estandarizadas. La de mayor reconocimiento y la más empleada es la publicada en 2015 por The American College of Medical Genetics and Genomics en colaboración con la Association for Molecular Pathology (ACMG/AMP)65.
Esta guía sienta las bases de la clasificación de las variantes genéticas en cinco grupos: patogénicas, probablemente patogénicas, de significado incierto, probablemente benignas y benignas. Para su clasificación en un grupo u otro se aplica un conjunto estructurado de criterios que pretende integrar la información conocida sobre las variantes: frecuencia poblacional, estudios funcionales, individuos previamente descritos en la literatura, tipo de impacto sobre la proteína codificada, segregación familiar, etc. Para cada uno de estos criterios, que se dividen en criterios de patogenicidad y de benignidad, se contempla una graduación a la que corresponde una puntuación: muy fuerte (8), fuerte (4), moderado (2) y de apoyo (1)66. Así, finalmente, tras la aplicar cada criterio con su correspondiente graduación, se obtiene una puntuación final que permite asignar una clasificación a la variante de interés (Tabla 2).
Tabla 2. Puntuaciones y probabilidad posterior de causar enfermedad para cada categoría de variante genética
| Categoría | Puntación | PP |
|---|---|---|
| Patogénica | ≥ 10 | > 99% |
| Probablemente patogénica | 6 a 9 | 99% ≥ PP > 90% |
| Significado incierto | 0 a 5 | 10% ≤ PP ≤ 90% |
| Probablemente benigna | –1 a –6 | 0,1% ≤ PP < 1% |
| Benigna | ≤ –7 | < 0,1% |
|
PP: probabilidad posterior. |
||
Este sistema define un marco de umbrales de probabilidad posterior que permite estimar la probabilidad de que una variante sea realmente causante de enfermedad. Así, las variantes clasificadas como patogénicas presentan una probabilidad > 99% de ser causales, mientras que las probablemente patogénicas se sitúan entre el 90-99%. De forma análoga, las variantes benignas presentan una probabilidad < 0,1% de causar enfermedad y las probablemente benignas entre el 0,1-10% (Tabla 2).
La guía del ACMG/AMP ha sido complementada por el grupo de trabajo Variant Interpretation (SVI) de ClinGen, y por la Association for Clinical Genomic Science (ACGS)67, lo que ha permitido refinar la aplicación de los diferentes criterios en la práctica clínica. Asimismo, también existen guías específicas de genes concretos que han desarrollado paneles de expertos de ClinGen, como ocurre con los diferentes genes de canales de sodio asociados a epilepsias: SCN1A, SCN1B, SCN2A, SCN3A y SCN8A.
Existen, a su vez, guías específicas para la interpretación y clasificación de variantes de tipo CNV68,69. Estas se basan en la misma lógica y determinan su clasificación dentro de las cinco categorías de patogenicidad conocidas.
En las enfermedades causadas por expansiones de repeticiones en tándem, las variantes se clasifican según el número de repeticiones detectadas en generalmente tres rangos: rango normal, no patogénico; rango intermedio (o premutación), caracterizado por inestabilidad meiótica y riesgo de expansión en las siguientes generaciones, pero sin asociación con la clínica; y rango patogénico, a partir del cual la expansión se asocia con manifestaciones clínicas. Una revisión sobre las enfermedades asociadas a expansiones, en la que se detallan los rangos para cada una de ellas, puede encontrarse en el siguiente trabajo70.
Resultados de los estudios genéticos
En los estudios genéticos esperamos tres tipos de resultados principales, que se explican a continuación.
RESULTADO POSITIVO
Se identifican variantes patogénicas y/o probablemente patogénicas, compatibles con el patrón de herencia esperado y que explican el fenotipo del paciente.
Cuando las variantes presentan valor clínico predictivo, se debe ofrecer el estudio genético en cascada a los familiares susceptibles de ser portadores. Cabe mencionar que para el estudio de individuos asintomáticos en enfermedades neurodegenerativas se recomienda un protocolo de asesoramiento genético específico71.
RESULTADO NEGATIVO
No se identifican variantes candidatas que puedan explicar el fenotipo del paciente. Un resultado negativo no excluye que la causa sea genética, ya que existen limitaciones técnicas y de conocimiento. Ante esta situación, es necesario valorar cuáles son las limitaciones específicas de la técnica utilizada y considerar la posibilidad de ampliar el estudio (véase el apartado de consideraciones específicas en cada trastorno).
Asimismo, tras un resultado negativo se puede plantear el reanálisis de los datos obtenidos mediante NGS, estrategia que ha demostrado incrementar el rendimiento diagnóstico cuando se realiza a partir de 1 a 3 años después del análisis inicial, debido principalmente a la incorporación de nuevos conocimientos científicos, la identificación de nuevas asociaciones gen-enfermedad y la mejora de las herramientas bioinformáticas de análisis, entre otros72. Aunque se ha planteado la posibilidad de realizar reanálisis sistemáticos de forma periódica, actualmente esto no suele estar implementado por lo que, en la mayoría de los casos, debe ser solicitado por el clínico responsable.
RESULTADO NO CONCLUYENTE
Se identifican variantes de significado incierto (VUS, variants of uncertain significance). Con las evidencias disponibles, no es posible ni confirmar ni descartar su relación causal con enfermedad, por lo que no deben tenerse en cuenta en la toma de decisiones clínicas. Esta situación es uno de los mayores retos en el diagnóstico genético.
Las VUS se presentan con un abanico amplio de probabilidad de causar enfermedad de entre el 10 y el 90%. Por tanto, no todas las VUS tienen el mismo grado de sospecha de patogenicidad. Aunque depende del laboratorio, la recomendación general es informar únicamente aquellas VUS susceptibles de reclasificación con la obtención de nuevas evidencias.
Esto puede lograrse mediante estudios de segregación familiar, tanto en casos esporádicos, cuando se demuestra el origen de novo de la variante, como en casos familiares, cuando puede establecerse su cosegregación con la enfermedad. Asimismo, puede ser útil la realización de pruebas complementarias dirigidas en determinadas enfermedades o genes con hallazgos específicos (marcadores bioquímicos, hallazgos característicos en estudios de imagen o fenotipos específicos). En algunos casos, los estudios funcionales, generalmente in vitro, pueden contribuir a esclarecer el efecto deletéreo de una variante, aunque habitualmente son complejos y se realizan en contexto de investigación. En variantes con potencial afectación al proceso de splicing pueden realizarse estudios de secuenciación de ARN. Cuando el gen se expresa en sangre periférica, esta aproximación puede ser relativamente accesible. También es posible la reclasificación de variantes en base a nuevas evidencias reportadas en la literatura o en las bases de datos clínicas o poblacionales.
HALLAZGOS INCIDENTALES Y HALLAZGOS SECUNDARIOS
En los estudios genéticos en los que se emplean técnicas de secuenciación masiva es habitual identificar hallazgos incidentales, definidos como aquellos que se detectan de forma no intencionada durante el análisis y que no están relacionados con el motivo clínico del estudio, y que pueden tener o no implicaciones para la salud. No existe un claro consenso de las sociedades científicas sobre si deben revelarse a los pacientes. Desde la European Society of Human Genetics (ESHG) se recomienda, al menos, que cada institución disponga de una política clara sobre su manejo y que esta sea explicada al paciente. Además, apuntan que debería considerarse su comunicación cuando indiquen un riesgo significativo para la salud del paciente o de sus familiares, especialmente cuando existan intervenciones preventivas o terapéuticas eficaces. Asimismo, algunos expertos y sociedades sostienen que debería ofrecerse a los pacientes la posibilidad de decidir, en el marco del consentimiento informado, si desean conocer este tipo de hallazgos73.
Los hallazgos secundarios son variantes genéticas identificadas de forma intencionada durante el análisis genómico que no están relacionadas con la indicación clínica inicial, pero se buscan de forma activa porque se considera que su detección es clínicamente accionable (existen intervenciones médicas capaces de prevenir o reducir significativamente la morbilidad y/o la mortalidad asociadas). La ACMG recomienda ofrecer su análisis en cualquier estudio de WES/WGS dentro del marco del consentimiento informado, en el contexto de un cribado oportunista. Para ello, propone una lista de genes actualizada periódicamente (84 genes en la versión más reciente, v3.3), asociados principalmente con predisposición al cáncer y enfermedades cardiovasculares, de los que únicamente se informan las variantes patogénicas o probablemente patogénicas74. La prevalencia estimada de estos hallazgos se sitúa aproximadamente entorno al 1-6%. Por su parte, la ESHG recomendaba en 2021 una aproximación prudente y progresiva a su implementación debido a la evidencia aún limitada sobre su beneficio clínico e impacto psicosocial73.
Conclusiones
El estudio genético se ha consolidado en el campo de la neurología como una herramienta clave en la práctica clínica, con impacto directo en el diagnóstico, manejo clínico y asesoramiento genético del paciente. Además, la incorporación progresiva de terapias dirigidas frente a alteraciones moleculares concretas refuerza el papel del estudio genético como un elemento esencial en el manejo del paciente.
Actualmente, la selección adecuada de los pacientes candidatos a estudios genéticos debe centrarse en aquellos en los que exista una sospecha razonable de causa monogénica, por lo que resulta fundamental conocer las indicaciones específicas en los distintos grupos de trastornos neurológicos.
La aplicación de las técnicas de NGS constituye actualmente un abordaje apropiado y eficiente para el estudio de la mayoría de estas enfermedades. No obstante, es importante considerar las limitaciones propias de cada técnica y reconocer aquellas situaciones en las que es necesario o más eficiente recurrir a otras técnicas específicas.
La complejidad en la interpretación de los resultados, junto con los retos derivados de la identificación de hallazgos incidentales o secundarios, hace imprescindible una colaboración estrecha entre genetistas y neurólogos, así como asegurar un adecuado asesoramiento genético a los pacientes en todas las fases del proceso diagnóstico.
Financiación
El presente trabajo no ha recibido ninguna subvención oficial, beca o apoyo de un programa de investigación destinados a la redacción de su contenido.
Conflicto de intereses
Los autores están empleados por Healthincode.
Consideraciones éticas
Protección de personas y animales. Los autores declaran que para este trabajo no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad, consentimiento informado y aprobación ética. El estudio no involucra datos personales de pacientes ni requiere aprobación ética. No se aplican las guías SAGER.
Declaración sobre el uso de inteligencia artificial. Los autores declaran que no utilizaron ningún tipo de inteligencia artificial generativa para la redacción de este manuscrito.
